Capítulo 6
Los elementos
La tabla periódica
Hasta ahora me he dedicado a los cuerpos del Universo de cierta entidad: las estrellas y galaxias, el Sistema Solar y la Tierra y su atmósfera. Ahora permítaseme considerar la naturaleza de las sustancias que componen todo esto.
Primeras teorías
Los primeros filósofos griegos, cuyo método de planteamiento de la mayor parte de los problemas era teórico y especulativo, llegaron a la conclusión de que la Tierra estaba formada por unos cuantos «elementos» o sustancias básicas. Empédocles de Agrigento, alrededor del 430 a. del J.C., estableció que tales elementos eran cuatro: tierra, aire, agua y fuego. Un siglo más tarde, Aristóteles supuso que el cielo constituía un quinto elemento: el «éter». Los sucesores de los griegos en el estudio de la materia, los alquimistas medievales, aunque sumergidos en la magia y la charlatanería, llegaron a conclusiones más razonables y verosímiles que las de aquéllos, ya que por lo menos manejaron los materiales sobre los que especulaban.
Tratando de explicar las diversas propiedades de las sustancias, los alquimistas atribuyeron dichas propiedades a determinados elementos, que añadieron a la lista. Identificaron el mercurio como el elemento que confería propiedades metálicas a las sustancias, y el azufre, como el que impartía la propiedad de la combustibilidad. Uno de los últimos y mejores alquimistas, el físico suizo del siglo XVI Theophrastus Bombasí von Hohenheim —más conocido por Paracelso—, añadió la sal como el elemento que confería a los cuerpos su resistencia al calor.
Según aquellos alquimistas, una sustancia puede transformarse en otra simplemente añadiendo y sustrayendo elementos en las proporciones adecuadas. Un metal como el plomo, por ejemplo, podía transformarse en oro añadiéndole una cantidad exacta de mercurio. Durante siglos prosiguió la búsqueda de la técnica adecuada para convertir en oro un «metal base». En este proceso, los alquimistas descubrieron sustancias mucho más importantes que el oro, tales como los ácidos minerales y el fósforo.
Los ácidos minerales —nítrico, clorhídrico y, especialmente, sulfúrico— introdujeron una verdadera revolución en los experimentos de la alquimia. Estas sustancias eran ácidos mucho más fuertes que el más fuerte conocido hasta entonces (el ácido acético, o sea, el del vinagre), y con ellos podían descomponerse las sustancias, sin necesidad de emplear altas temperaturas ni recurrir a largos períodos de espera. Aún en la actualidad, los ácidos minerales, especialmente el sulfúrico, son muy importantes en la industria. Se dice incluso que el grado de industrialización de un país puede ser juzgado por su consumo anual de ácido sulfúrico.
De todas formas, pocos alquimistas se dejaron tentar por estos importantes éxitos secundarios, para desviarse de lo que ellos consideraban su búsqueda principal. Sin embargo, miembros poco escrupulosos de la profesión llegaron abiertamente a la estafa, simulando, mediante juegos de prestidigitación, producir oro, al objeto de conseguir lo que hoy llamaríamos «becas para la investigación» por parte de ricos mecenas. Este arte consiguió así tan mala reputación, que hasta la palabra «alquimista» tuvo que ser abandonada. En el siglo XVII, «alquimista» se había convertido en «químico», y «alquimia» había pasado a ser la ciencia llamada «Química».
En el brillante nacimiento de esta ciencia, uno de los primeros genios fue Robert Boyle, quien formuló la ley de los gases que hoy lleva su nombre (véase capítulo 5). En su obra El químico escéptico (The Sceptical Chymist), publicada en 1661, Boyle fue el primero en establecer el criterio moderno por el que se define un elemento: una sustancia básica que puede combinarse con otros elementos para formar «compuestos» y que, por el contrario, no puede descomponerse en una sustancia más simple, una vez aislada de un compuesto.
Sin embargo, Boyle conservaba aún cierta perspectiva medieval acerca de la naturaleza de los elementos. Por ejemplo, creía que el oro no era un elemento y que podía formarse de algún modo a partir de otros metales. Las mismas ideas compartía su contemporáneo Isaac Newton, quien dedicó gran parte de su vida a la alquimia. (En realidad, el emperador Francisco José de Austria-Hungría financió experimentos para la fabricación de oro hasta fecha tan reciente como 1867.)
Un siglo después de Boyle, los trabajos prácticos realizados por los químicos empezaron a poner de manifiesto qué sustancias podrían descomponerse en otras más simples y cuáles no podían ser descompuestas. Henry Cavendish demostró que el hidrógeno se combinaba con el oxígeno para formar agua, de modo que ésta no podía ser un elemento. Más tarde, Lavoisier descompuso el aire —que se suponía entonces un elemento— en oxígeno y nitrógeno. Se hizo evidente que ninguno de los «elementos» de los griegos eran tales según el criterio de Boyle.
En cuanto a los elementos de los alquimistas, el mercurio y el azufre resultaron serlo en el sentido de Boyle. Y también lo eran el hierro, el estaño, el plomo, el cobre, la plata, el oro y otros no metálicos, como el fósforo, el carbono y el arsénico. El «elemento» de Paracelso (la sal) fue descompuesto en dos sustancias más simples.
Desde luego, el que un elemento fuera definido como tal dependía del desarrollo alcanzado por la Química en la época. Mientras una sustancia no pudiera descomponerse con ayuda de las técnicas químicas disponibles, debía seguir siendo considerada como un elemento. Por ejemplo, la lista de 33 elementos formulada por Lavoisier incluía, entre otros, los óxidos de cal y magnesio. Pero catorce años después de la muerte de Lavoisier en la guillotina, durante la Revolución francesa, el químico inglés Humphry Davy, empleando una corriente eléctrica para escindir las sustancias, descompuso la cal en oxígeno y en un nuevo elemento, que denominó «calcio»; luego escindió el óxido de magnesio en oxígeno y otro nuevo elemento, al que dio el nombre de «magnesio».
Por otra parte, Davy demostró que el gas verde obtenido por el químico sueco Cari Wilhelm Scheele a partir del ácido clorhídrico no era un compuesto de ácido clorhídrico y oxígeno, como se había supuesto, sino un verdadero elemento, al que denominó «cloro» (del griego cloros, verde amarillento).
Teoría atómica
A principios del siglo XIX, el químico inglés John Dalton contempló los elementos desde un punto de vista totalmente nuevo. Por extraño que parezca, esta perspectiva se remonta, en cierto modo, a la época de los griegos, quienes, después de todo, contribuyeron con lo que tal vez sea el concepto simple más importante para la comprensión de la materia.
Los griegos se planteaban la cuestión de si la materia era continua o discontinua, es decir, si podía ser dividida y subdividida indefinidamente en un polvo cada vez más fino, o si, al término de este proceso se llegaría a un punto en el que las partículas fuesen indivisibles. Leucipo de Mileto y su discípulo Demócrito de Abdera insistían —en el año 450 a. de J.C.— en que la segunda hipótesis era la verdadera. Demócrito dio a estas partículas un nombre: las llamó «átomos» (o sea, «no divisibles»). Llegó incluso a sugerir que algunas sustancias estaban compuestas por diversos átomos o combinaciones de átomos, y que una sustancia podría convertirse en otra al ordenar dichos átomos de forma distinta. Si tenemos en cuenta que esto es sólo una sutil hipótesis, no podemos por menos que sorprendernos ante la exactitud de su intuición. Pese a que la idea pueda parecer hoy evidente, estaba muy lejos de serlo en la época en que Platón y Aristóteles la rechazaron.
Sin embargo, sobrevivió en las enseñanzas de Epicuro de Samos —quien escribió sus obras hacia el año 300 a. de J.C.— y en la escuela filosófica creada por él: el epicureísmo. Un importante epicúreo fue el filósofo romano Lucrecio, quien, sobre el año 60 a. de J.C., plasmó sus ideas acerca del átomo en un largo poema titulado Sobre la naturaleza de las cosas. Este poema sobrevivió a través de la Edad Media y fue uno de los primeros trabajos que se imprimieron cuando lo hizo posible el arte de Gutenberg.
La noción de los átomos nunca fue descartada por completo de las escuelas occidentales. Entre los atomistas más destacados en los inicios de la Ciencia moderna figuran el filósofo italiano Giordano Bruno y el filósofo francés Pierre Gassendi. Muchos puntos de vista científicos de Bruno no eran ortodoxos, tales como la creencia en un Universo infinito sembrado de estrellas, que serían soles lejanos, alrededor de los cuales evolucionarían planetas, y expresó temerariamente sus teorías. Fue quemado, por hereje, en 1600, lo cual hizo de él un mártir de la Ciencia en la época de la revolución científica. Los rusos han dado su nombre a un cráter de la cara oculta de la Luna.
Las teorías de Gassendi impresionaron a Boyle, cuyos experimentos, reveladores de que los gases podían ser fácilmente comprimidos y expandidos, parecían demostrar que estos gases debían de estar compuestos por partículas muy espaciadas entre sí. Por otra parte, tanto Boyle como Newton figuraron entre los atomistas más convencidos del siglo XVII.
En 1799, el químico francés Joseph Louis Proust mostró que el carbonato de cobre contenía unas proporciones definidas de peso de cobre, carbono y oxígeno y que podía prepararse. Las proporciones seguían el índice de unos pequeños números enteros: 5 a 4 y a 1. Demostró que existía una situación similar para cierto número de otros compuestos.
Esta situación podía explicarse dando por supuesto que los compuestos estaban formados por la unión de pequeños números de fragmentos de cada elemento y que sólo podían combinarse como objetos intactos. El químico inglés John Dalton señaló todo esto en 1803, y, en 1808, publicó un libro en el que se reunía la nueva información química conseguida durante el siglo y medio anterior, y que sólo tenía sentido si se suponía que la materia estaba compuesta de átomos indivisibles. (Dalton mantuvo la antigua voz griega como tributo a los pensadores de la Antigüedad.) No pasó mucho tiempo antes de que esta teoría atómica persuadiera a la mayoría de los químicos. Según Dalton, cada elemento posee una clase particular de átomo, y cualquier cantidad de elemento está compuesta de átomos idénticos de esa clase. Lo que distingue a un elemento de otro es la naturaleza de sus átomos. Y la diferencia física básica entre los átomos radica en su peso. Así, los átomos de azufre son más pesados que los de oxígeno, que, a su vez, son más pesados que los átomos de nitrógeno; éstos, a su vez también, son más pesados que los de carbono, y los mismos, más pesados que los de hidrógeno.
El químico italiano Amadeo Avogadro aplicó a los gases la teoría atómica y demostró que volúmenes iguales de un gas, fuese cual fuese su naturaleza, estaban formados por el mismo número de partículas. Es la llamada «hipótesis de Avogadro». Al principio se creyó que estas partículas eran átomos; pero luego se demostró que estaban compuestas, en la mayor parte de los casos, por pequeños grupos de átomos, llamados «moléculas». Si una molécula contiene átomos de distintas clases (como la molécula de agua, que tiene un átomo de oxígeno y dos de hidrógeno), es una molécula de un «compuesto químico». Naturalmente, era importante medir los pesos relativos de los distintos átomos, para hallar los «pesos atómicos» de los elementos. Pero los pequeños átomos se hallaban muy lejos de las posibilidades ponderables del siglo XIX. Mas, pesando la cantidad de cada elemento separado de un compuesto y haciendo deducciones a partir del comportamiento químico de los elementos, se pudieron establecer los pesos relativos de los átomos. El primero en realizar este trabajo de forma sistemática fue el químico sueco Jóns Jacob Berzelius. En 1828 publicó una lista de pesos atómicos basados en dos patrones de referencia: uno, el obtenido al dar el peso atómico del oxígeno el valor 100, y el otro, cuando el peso atómico del hidrógeno se hacía igual a 1.
El sistema de Berzelius no alcanzó inmediata aceptación; pero en 1860, en el I Congreso Internacional de Química, celebrado en Karlsruhe (Alemania), el químico italiano Stanislao Cannizzaro presentó nuevos métodos para determinar los pesos atómicos, con ayuda de la hipótesis de Avogadro, menospreciada hasta entonces. Describió sus teorías de forma tan convincente, que el mundo de la Química quedó conquistado inmediatamente. Se adoptó como unidad de medida el peso del oxígeno en vez del del hidrógeno, puesto que el oxígeno podía ser combinado más fácilmente con los diversos elementos —y tal combinación era el punto clave del método usual para determinar los pesos atómicos—. El peso atómico del oxígeno fue medido convencionalmente, en 1850, por el químico belga Jean Serváis Stas, quien lo fijó en 16, de modo que el peso atómico del hidrógeno, el elemento más ligero conocido hasta ahora, sería, aproximadamente, de 1; para ser más exactos: 1,0080.
Desde la época de Cannizzaro, los químicos han intentado determinar los pesos atómicos cada vez con mayor exactitud. Por lo que se refiere a los métodos puramente químicos, se llegó al punto culminante con los trabajos del químico norteamericano Theodore William Richards, quien, desde 1904, se dedicó a determinar los pesos atómicos con una exactitud jamás alcanzada. Por ello se le concedió el premio Nobel de Química en 1914. En virtud de los últimos descubrimientos sobre la constitución física de los átomos, las cifras de Richards han sido corregidas desde entonces y se les han dado valores aún más exactos. A lo largo del siglo XIX y pese a realizar múltiples investigaciones que implicaban la aceptación de las nociones de átomos y moléculas y a que, por lo general, los científicos estaban convencidos de su existencia, no se pudo aportar ninguna prueba directa de que fuesen algo más que simples abstracciones convenientes. Algunos destacados científicos, como el químico alemán Wilhelm Ostwald, se negaron a aceptarlos. Para él eran conceptos útiles, pero no «reales».
La existencia real de las moléculas la puso de manifiesto el «movimiento browniano», que observó por vez primera, en 1827, el botánico escocés Robert Brown, quien comprobó que los granos de polen suspendidos en el agua aparecían animados de movimientos erráticos. Al principio se creyó que ello se debía a que los granos de polen tenían vida; pero, de forma similar, se observó que también mostraban movimiento pequeñas partículas de sustancias colorantes totalmente inanimadas.
En 1863 se sugirió por vez primera que tal movimiento sería debido a un bombardeo desigual de las partículas por las moléculas de agua circundantes. En los objetos macroscópicos no tendría importancia una pequeña desigualdad en el número de moléculas que incidieran de un lado u otro. Pero en los objetos microscópicos, bombardeados quizá por sólo unos pocos centenares de moléculas por segundo, un pequeño exceso —por uno u otro lado— podría determinar una agitación perceptible. El movimiento al azar de las pequeñas partículas constituye una prueba casi visible de que el agua, y la materia en general, tiene «partículas».
Einstein elaboró un análisis teórico del movimiento browniano y demostró cómo se podía averiguar el tamaño de las moléculas de agua considerando la magnitud de los pequeños movimientos en zigzag de las partículas de colorantes. En 1908, el científico francés Jean Perrin estudió la forma en que las partículas se posaban, como sedimento, en el agua, debido a la influencia de la gravedad. A esta sedimentación se oponían las colisiones determinadas por las moléculas procedentes de niveles inferiores, de modo que el movimiento browniano se oponía a la fuerza gravitatoria. Perrin utilizó este descubrimiento para calcular el tamaño de las moléculas de agua mediante la ecuación formulada por Einstein, e incluso Oswald tuvo que ceder en su postura. Estas investigaciones le valieron a Perrin, en 1926, el premio Nobel de Física.
Así, pues, los átomos se convirtieron, de abstracciones semimísticas, en objetos casi tangibles. En realidad, hoy podemos decir que, al fin, el hombre ha logrado «ver» el átomo. Ello se consigue con el llamado «microscopio de campo iónico», inventado, en 1955, por Erwin W. Mueller, de la Universidad de Pensilvania. El aparato arranca iones de carga positiva a partir de la punta de una aguja finísima, iones que inciden contra una pantalla fluorescente, la cual da una imagen, ampliada 5 millones de veces, de la punta de la aguja. Esta imagen permite que se vea como un pequeño puntito brillante cada uno de los átomos que componen la punta. La técnica alcanzaría su máxima perfección cuando pudieran obtenerse imágenes de cada uno de los átomos por separado. En 1970, el físico americano Albert Víctor Crewe informó que había detectado átomos sueltos de uranio y torio con ayuda del microscopio electrónico.
La tabla periódica de Mendeléiev
A medida que, durante el siglo XIX, fue aumentando la lista de los elementos, los químicos empezaron a verse envueltos en una intrincada maleza. Cada elemento tenía propiedades distintas, y no daban con ninguna fórmula que permitiera ordenar aquella serie de elementos. Puesto que la Ciencia tiene como finalidad el tratar de hallar un orden en un aparente desorden, los científicos buscaron la posible existencia de caracteres semejantes en las propiedades de los elementos.
En 1862, después de haber establecido Cannizzaro el peso atómico como una de las más importantes herramientas de trabajo de la Química, un geólogo francés (Alexandre-Émile Beguyer de Chancourtois) comprobó que los elementos se podían disponer en forma de tabla por orden creciente, según su peso atómico, de forma que los de propiedades similares se hallaran en la misma columna vertical. Dos años más tarde, un químico británico (John Alexander Reina Newlands) llegó a disponerlos del mismo modo, independientemente de Beguyer. Pero ambos científicos fueron ignorados o ridiculizados. Ninguno de los dos logró ver impresas sus hipótesis. Muchos años más tarde, una vez reconocida universalmente la importancia de la tabla periódica, sus investigaciones fueron publicadas al fin. A Newlands se le concedió incluso una medalla.
El químico ruso Dmitri Ivánovich Mendeléiev fue reconocido, finalmente, como el investigador que puso orden en la selva de los elementos. En 1869, él y el químico alemán Julius Lothar Meyer propusieron tablas de los elementos que, esencialmente, se regían por las ideas de Chancourtois y Newlands. Pero Mendeléiev fue reconocido por la Ciencia, porque tuvo el valor y la confianza de llevar sus ideas más allá que los otros.
En primer lugar, la tabla periódica «de Mendeléiev» —llamada «periódica» porque demostraba la repetición periódica de propiedades químicas similares— era más complicada que la de Newlands y más parecida a la que hoy estimamos como correcta (fig. 6.1). En segundo lugar, cuando las propiedades de un elemento eran causa de que no conservara el orden establecido en función de su peso atómico, cambiaba resueltamente el orden, basándose en que las propiedades eran más importantes que el peso atómico. Se demostró que ello era correcto. Por ejemplo, el telurio, con un peso atómico de 127,60, debería estar situado, en función de los pesos, después del yodo, cuyo peso atómico es de 126,90; pero en la tabla dispuesta en columnas, cuando se coloca el telurio delante del yodo, se halla bajo el selenio, que se asemeja mucho a él, y, del mismo modo, el yodo aparece debajo de su afín, el bromo.
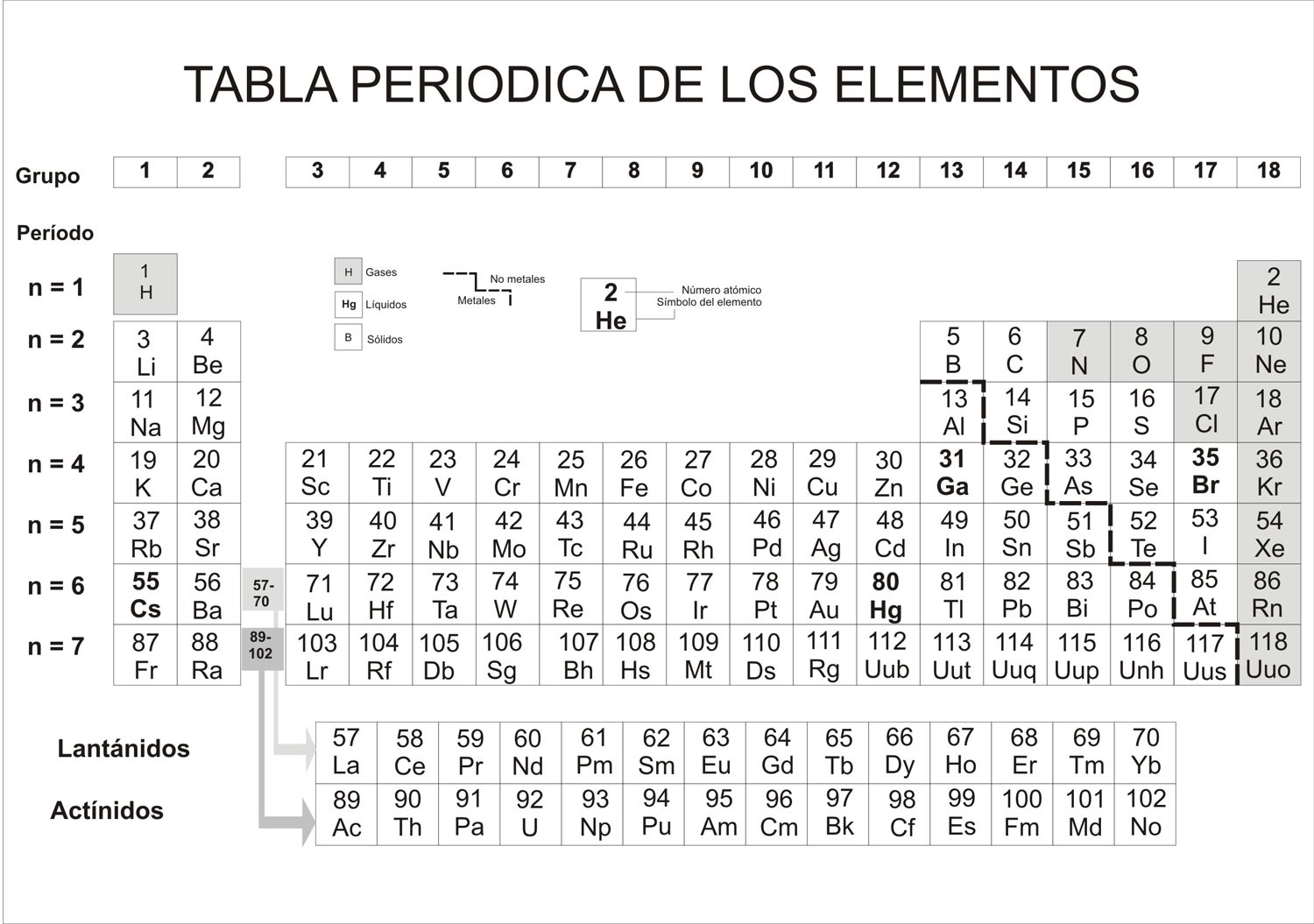
Fig. 6.1. Tabla periódica de los elementos.
Finalmente —y esto es lo más importante—, cuando Mendeléiev no conseguía que los elementos encajaran bien en el sistema, no vacilaba en dejar espacios vacíos en la tabla y anunciar —con lo que parecía un gran descaro— que faltaban por descubrir elementos, los cuales rellenarían los vacíos. Pero fue aún más lejos. Describió el elemento que correspondía a cada uno de tres vacíos, utilizando como guía las propiedades de los elementos situados por encima y por debajo del vacío en la tabla. Aquí, Mendeléiev mostróse genialmente intuitivo. Los tres elementos predichos fueron encontrados, ya en vida de Mendeléiev, por lo cual pudo asistir al triunfo de su sistema. En 1875, el químico francés Lecoq de Boisbaudran descubrió el primero de dichos elementos, al que denominó «galio» (del latín gallium, Francia). En 1879, el químico sueco Lars Fredrik Nilson encontró el segundo, y lo llamó «escandio» (por Escandinavia). Y en 1886, el químico alemán Clemens Alexander Winkler aisló el tercero y lo denominó «germanio» (naturalmente, por Germania). Los tres elementos mostraban casi las mismas propiedades que predijera Mendeléiev.
Números atómicos
Con el descubrimiento de los rayos X[2] se abrió una nueva Era en la historia de la tabla periódica. En 1911, el físico británico Charles Glover Barkla descubrió que cuando los rayos X se dispersaban al atravesar un metal, dichos rayos, refractados, tenían un sensible poder de penetración, que dependía de la naturaleza del metal. En otras palabras, que cada elemento producía sus «rayos X característicos». Por este descubrimiento, Barkla fue galardonado con el premio Nobel de Física en 1917.
Existían algunas dudas sobre si los rayos X eran corrientes de pequeñas partículas o consistían en radiaciones de carácter ondulatorio similares, en este sentido, a la luz. Una manera de averiguarlo era el comprobar si los rayos X podían ser difractados (es decir, forzados a cambiar de dirección) mediante un dispositivo difractante, constituido por una serie de finas líneas paralelas. Sin embargo, para una difracción adecuada, la distancia entre las líneas debe ser igual al tamaño de las ondas de la radiación. El conjunto de líneas más tupido que podía prepararse era suficiente para la luz ordinaria; pero el poder de penetración de los rayos X permitía suponer como probable —admitiendo que dichos rayos fuesen de naturaleza ondulatoria— que las ondas eran mucho más pequeñas que las de la luz. Por tanto, ningún dispositivo de difracción usual bastaba para difractar los rayos X.
Sin embargo, el físico alemán Max Theodore Félix von Laue observó que los cristales constituían una retícula de difracción natural mucho más fina que cualquiera de los fabricados por el hombre. Un cristal es un cuerpo sólido de forma claramente geométrica, cuyas caras planas se cortan en ángulos determinados, de simetría característica. Esta visible regularidad es el resultado de una ordenada disposición de los átomos que forman su estructura. Había razones para creer que el espacio entre una capa de átomos y la siguiente tenía, aproximadamente, las dimensiones de una longitud de onda de los rayos X. De ser así, los cristales difractarían los rayos X.
En sus experimentos, Laue comprobó que los rayos X que pasaban a través de un cristal eran realmente difractados y formaban una imagen sobre una placa fotográfica, que ponía de manifiesto su carácter ondulatorio. En el mismo año, el físico inglés William Lawrence Bragg y su padre, William Henry Bragg, desarrollaron un método exacto para calcular la longitud de onda de un determinado tipo de rayos X, a partir de su imagen de difracción. A la inversa, se emplearon imágenes de difracción de rayos X para determinar la orientación exacta de las capas de átomos que causaban su difracción. De este modo, los rayos X abrieron la puerta a una nueva comprensión de la estructura atómica de los cristales. Por su trabajo sobre los rayos X, Laue recibió el premio Nobel de Física en 1914, mientras que los Bragg lo compartieron en 1915.
En 1914, el joven físico inglés Henry Gwyn-Jeffreys Moseley determinó las longitudes de onda de los característicos rayos X producidos por diversos metales, e hizo el importante descubrimiento de que la longitud de onda disminuía de forma regular al avanzar en la tabla periódica.
Ello permitió situar de manera definitiva los elementos en la tabla. Si dos elementos, supuestamente adyacentes en la tabla, emitían rayos X cuyas longitudes de onda diferían en una magnitud doble de la esperada, debía de existir un vacío entre ellos, perteneciente a un elemento desconocido. Si diferían en una magnitud tres veces superior a la esperada, debían de existir entre ellos dos elementos desconocidos. Si, por otra parte, las longitudes de onda de los rayos X característicos de los dos elementos diferían sólo en el valor esperado, podía tenerse la seguridad de que no existía ningún elemento por descubrir entre los otros dos.
Por tanto, se podía dar números definitivos a los elementos. Hasta entonces había cabido siempre la posibilidad de que un nuevo descubrimiento rompiera la secuencia y trastornara cualquier sistema de numeración adoptado. Ahora ya no podían existir vacíos inesperados.
Los químicos procedieron a numerar los elementos desde el 1 (hidrógeno) hasta el 92 (uranio). Estos «números atómicos» resultaron significativos en relación con la estructura interna de los átomos (véase capítulo 7), y de una importancia más fundamental que el peso atómico. Por ejemplo, los datos proporcionados por los rayos X demostraron que Mendeléiev había tenido razón al colocar el telurio (de número atómico 52) antes del yodo (53), pese a ser mayor el peso atómico del telurio.
El nuevo sistema de Moseley demostró su valor casi inmediatamente. El químico francés Georges Urbain, tras descubrir el «lutecio» (por el nombre latino de París, Lutecia), anunció que acababa de descubrir otro elemento, al que llamó «celtio». De acuerdo con el sistema de Moseley, el lutecio era el elemento 71, y el celtio debía ser el 72. Pero cuando Moseley analizó los rayos X característicos del celtio, resultó que se trataba del mismo lutecio. El elemento 72 no fue descubierto realmente hasta 1923, cuando el físico danés Dirk Coster y el químico húngaro Georg von Hevesy lo detectaron en un laboratorio de Copenhague. Lo denominaron «hafnio», por el nombre latinizado de Copenhague.
Pero Moseley no pudo comprobar la exactitud de su método, pues había muerto en Gallípoli, en 1915, a los veintiocho años de edad. Fue uno de los cerebros más valiosos perdidos en la Primera Guerra Mundial. Ello le privó también, sin duda, del premio Nobel. El físico sueco Karl Manne George Siegbahn amplió el trabajo de Moseley, al descubrir nuevas series de rayos X y determinar con exactitud el espectro de rayos X de los distintos elementos. En 1924 fue recompensado con el premio Nobel de Física.
En 1925, Walter Noddack, Ida Tacke y Otto Berg, de Alemania, llenaron otro vacío en la tabla periódica. Después de treinta años de investigar los minerales que contenían elementos relacionados con el que estaban buscando, descubrieron el elemento 75, al que dieron el nombre de «renio», en honor al río Rin. De este modo se reducían a cuatro los espacios vacíos: correspondían a los elementos 43, 61, 85 y 87.
Fueron necesarias dos décadas para encontrarlos. A pesar de que los químicos de entonces no se percataron de ello, habían hallado el último de los elementos estables. Los que faltaban eran especies inestables tan raras hoy en la Tierra, que todas menos una tuvieron que ser creadas en el laboratorio para identificarlas. Y este descubrimiento va asociado a una historia.
Elementos radiactivos
Identificación de los elementos
Tras el descubrimiento de los rayos X, muchos científicos se sintieron impulsados a investigar estas nuevas radiaciones, tan espectacularmente penetrantes. Uno de ellos fue el físico francés Antoine-Henry Becquerel. El padre de Henry, Alexandre Edmond (el físico que fotografío por vez primera el espectro solar), se había mostrado especialmente interesado en la «fluorescencia», o sea, la radiación visible emitida por sustancias después de ser expuestas a los rayos ultravioletas de la luz solar.
Becquerel padre había estudiado, en particular, una sustancia fluorescente llamada sulfato de uranilo potásico (compuesto formado por moléculas, cada una de las cuales contiene un átomo de uranio). Henry se preguntó si las radiaciones fluorescentes del sulfato de uranilo potásico contenían rayos X. La forma de averiguarlo consistía en exponer el sulfato al Sol (cuya luz ultravioleta estimularía la fluorescencia), mientras el compuesto permanecía sobre una placa fotográfica envuelta en papel negro. Puesto que la luz solar no podía penetrar a través del papel negro, no afectaría a la placa; pero si la fluorescencia producida por el estímulo de la luz solar contenía rayos X, estos penetrarían a través del papel e impresionarían la placa. Becquerel realizó con éxito su experimento en 1896. Aparentemente, había rayos X en la fluorescencia. Becquerel logró incluso que los supuestos rayos X pasasen a través de delgadas láminas de aluminio y cobre, y los resultados parecieron confirmar definitivamente su hipótesis, puesto que no se conocía radiación alguna, excepto la de los rayos X, que pudiese hacerlo.
Pero entonces —lo cual fue una suerte— el Sol quedó oculto por densos nubarrones. Mientras esperaba que se disiparan las nubes, Becquerel retiró las placas fotográficas, con los trocitos de sulfato sobre ellas, y las puso en un secador. Al cabo de unos días, impaciente, decidió a toda costa revelar las placas, en la creencia de que, incluso sin la luz solar directa, se podía haber producido alguna pequeña cantidad de rayos X. Cuando vio las placas impresionadas, Becquerel vivió uno de esos momentos de profunda sorpresa y felicidad que son los sueños de todos los científicos. La placa fotográfica estaba muy oscurecida por una intensa radiación. La causa no podía ser la fluorescencia ni la luz solar. Bequerel llegó a la conclusión —y los experimentos lo confirmarían muy pronto— de que esta causa era el propio uranio contenido en el sulfato de uranilo potásico.
Este descubrimiento impresionó profundamente a los científicos, excitados aún por el reciente hallazgo de los rayos X. Uno de los científicos que se puso inmediatamente a investigar la extraña radiación del uranio fue una joven química, nacida en Polonia y llamada Marie Sklodowska, que el año anterior había contraído matrimonio con Pierre Curie, el descubridor de la temperatura que lleva su nombre (véase capítulo 5).
Pierre Curie, en colaboración con su hermano Jacques, había descubierto que ciertos cristales, sometidos a presión, desarrollaban una carga eléctrica positiva en un lado y negativa en el otro. Este fenómeno se denomina «piezoelectricidad» (de la voz griega que significa «comprimir»). Marie Curie decidió medir, con ayuda de la piezoelectricidad, la radiación emitida por el uranio. Instaló un dispositivo, al que fluiría una corriente cuando la radiación ionizase el aire entre dos electrodos; y la potencia de esta pequeña corriente podría medirse por la cantidad de presión que debía ejercerse sobre un cristal para producir una corriente contraria que la anulase. Este método se mostró tan efectivo, que Pierre Curie abandonó en seguida su trabajo, y durante el resto de su vida, junto con Marie, se dedicó a investigar ávidamente en este campo.
Marie Curie fue la que propuso el término de «radiactividad» para describir la capacidad que tiene el uranio de emitir radiaciones, y la que consiguió demostrar el fenómeno en una segunda sustancia radiactiva: el torio. En rápida sucesión, otros científicos hicieron descubrimientos de trascendental importancia. Las radiaciones de las sustancias radiactivas se mostraron más penetrantes y de mayor energía que los rayos X; hoy se llaman «rayos gamma». Se descubrió que los elementos radiactivos emitían también otros tipos de radiación, lo cual condujo a descubrimientos sobre la estructura interna del átomo. Pero esto lo veremos en el capítulo 7. Lo que nos interesa destacar aquí es el descubrimiento de que los elementos radiactivos, al emitir la radiación, se transformaban en otros elementos (o sea, era una versión moderna de la transmutación).
Marie Curie descubrió, aunque de forma accidental, las implicaciones de este fenómeno. Cuando ensayaba la pechblenda en busca de su contenido de uranio, al objeto de comprobar si las muestras de la mena tenían el uranio suficiente para hacer rentable la labor del refinado, ella y su marido descubrieron, con sorpresa, que algunos de los fragmentos tenían más radiactividad de la esperada, aunque estuviesen hechos de uranio puro. Ello significaba que en la pechblenda habían de hallarse otros elementos radiactivos, aunque sólo en pequeñas cantidades (oligoelementos), puesto que el análisis químico usual no los detectaba; pero, al mismo tiempo, debían ser muy radiactivos.
Entusiasmados, los Curie adquirieron toneladas de pechblenda, construyeron un pequeño laboratorio en un cobertizo y, en condiciones realmente primitivas, procedieron a desmenuzar la pesada y negra mena, en busca de los nuevos elementos. En julio de 1898 habían conseguido aislar un polvo negro 400 veces más radiactivo que la cantidad equivalente de uranio.
Este polvo contenía un nuevo elemento, de propiedades químicas parecidas a las del telurio, por lo cual debía colocarse bajo este en la tabla periódica. (Más tarde se le dio el número atómico 84.) Los Curie lo denominaron «polonio», en honor al país natal de Marie.
Pero el polonio justificaba sólo una parte de la radiactividad. Siguieron nuevos trabajos, y en diciembre de 1898, los Curie habían obtenido un preparado que era incluso más radiactivo que el polonio. Contenía otro elemento, de propiedades parecidas a las del bario (y, eventualmente, se puso debajo de éste, con el número atómico 88). Los Curie lo llamaron «radio», debido a su intensa radiactividad.
Siguieron trabajando durante cuatro años más, para obtener una cantidad de radio puro que pudiese apreciarse a simple vista. En 1903, Marie Curie presentó un resumen de su trabajo como tesis doctoral. Tal vez sea la mejor tesis de la historia de la Ciencia. Ello le supuso no sólo uno, sino dos premios Nobel. Marie y su marido, junto con Becquerel, recibieron, en 1903, el de Física, por sus estudios sobre la radiactividad, y, en 1911, Marie —su marido había muerto en 1906, en accidente de circulación— fue galardonada con el de Química por el descubrimiento del polonio y el radio.
El polonio y el radio son mucho más inestables que el uranio y el torio, lo cual es otra forma de decir que son mucho más radiactivos. En cada segundo se desintegra mayor número de sus átomos. Sus vidas son tan cortas, que prácticamente todo el polonio y el radio del Universo deberían haber desaparecido en un millón de años. Por tanto, ¿cómo seguimos encontrándolo en un planeta que tiene miles de millones de años de edad? La respuesta es que el radio y el polonio se van formando continuamente en el curso de la desintegración del uranio y el torio, para acabar por transformarse en plomo. Dondequiera que se hallen el uranio y el torio, se encuentran siempre indicios de polonio y radio. Son productos intermedios en el camino que conduce al plomo como producto final.
El detenido análisis de la pechblenda y las investigaciones de las sustancias radiactivas permitieron descubrir otros tres elementos inestables en el camino que va del uranio y el torio hasta el plomo. En 1899, André-Louis Debierne, siguiendo el consejo de los Curie, buscó otros elementos en la pechblenda y descubrió uno, al que denominó «actinio» (de la voz griega que significa «rayo»); se le dio el número atómico 89. Al año siguiente, el físico alemán Friedrich Ernst Dorn demostró que el radio, al desintegrarse, formaba un elemento gaseoso. ¡Un gas radiactivo era algo realmente nuevo! El elemento fue denominado «radón» (de radio y argón, su afín químico), y se le dio el número atómico 86. Finalmente, en 1917, dos grupos distintos —Otto Hahn y Lise Meitner, en Alemania, y Frederick Soddy y John A. Cranston, en Inglaterra— aislaron, a partir de la pechblenda, el elemento 91, denominado protactinio.
El hallazgo de los elementos perdidos
Por tanto, en 1925 había 88 elementos identificados: 81 estables, y 7, inestables. Se hizo más acuciante la búsqueda de los cuatro que aún faltaban: los números 43, 61, 85 y 87.
Puesto que entre los elementos conocidos había una serie radiactiva —los números 84 al 92—, podía esperarse que también lo fueran el 85 y el 87. Por otra parte, el 43 y el 61 estaban rodeados por elementos estables, y no parecía haber razón alguna para sospechar que no fueran, a su vez, estables. Por tanto, deberían de encontrarse en la Naturaleza.
Respecto al elemento 43, situado inmediatamente encima del reino en la tabla periódica, se esperaba que tuviese propiedades similares y que se encontrase en las mismas menas. De hecho, el equipo de Noddack, Tacke y Berg, que había descubierto el renio, estaba seguro de haber dado también con rayos X de una longitud de onda que debían de corresponder al elemento 43. Así, pues, anunciaron su descubrimiento, y lo denominaron «masurio» (por el nombre de una región de la Prusia Oriental). Sin embargo, su identificación no fue confirmada, y, en Ciencia, un descubrimiento no se considera como tal hasta que haya sido confirmado, como mínimo, por un investigador independiente.
En 1926, dos químicos de la Universidad de Illinois anunciaron que habían encontrado el elemento 61 en menas que contenían los elementos vecinos (60 y 62), y lo llamaron «illinio». El mismo año, dos químicos italianos de la Universidad de Florencia creyeron haber aislado el mismo elemento, que bautizaron con el nombre de «florencio». Pero el trabajo de ambos grupos no pudo ser confirmado por ningún otro.
Años más tarde, un físico del Instituto Politécnico de Alabama, utilizando un nuevo método analítico de su invención, informó haber encontrado indicios de los elementos 87 y 85, a los que llamó «virginio» y «alabaminio», en honor, respectivamente, de sus Estados natal y de adopción. Pero tampoco pudieron ser confirmados estos descubrimientos.
Los acontecimientos demostrarían que, en realidad, no se habían descubierto los elementos 43, 61, 85 y 87.
El primero en ser identificado con toda seguridad fue el elemento 43. El físico estadounidense Ernest Orlando Lawrence —quien más tarde recibiría el premio Nobel de Física como inventor del ciclotrón (véase capítulo 7)— obtuvo el elemento en su acelerador mediante el bombardeo de molibdeno (elemento 42) con partículas a alta velocidad. El material bombardeado mostraba radiactividad, y Lawrence lo remitió al químico italiano Emilio Gino Segré —quien estaba interesado en el elemento 43— para que lo analizase. Segré y su colega C. Perrier, tras separar la parte radiactiva del molibdeno, descubrieron que se parecía al renio en sus propiedades. Y decidieron que sólo podía ser el elemento 43, elemento que contrariamente a sus vecinos de la tabla periódica, era radiactivo. Al no ser producidos por desintegración de un elemento de mayor número atómico, apenas quedan indicios del mismo en la corteza terrestre, por lo cual, Noddack y su equipo estaban equivocados al creer que lo habían hallado. Segré y Perrier tuvieron el honor de bautizar el elemento 43; lo llamaron «tecnecio», tomado de la voz griega que significa «artificial», porque éste era el primer elemento fabricado por el hombre. Hacia 1960 se había acumulado ya el tecnecio suficiente para determinar su punto de fusión: cercano a los 2.200 °C. (Segré recibió posteriormente el premio Nobel por otro descubrimiento, relacionado también con materia creada por el hombre [véase capítulo 7].)
Finalmente, en 1939, se descubrió en la Naturaleza el elemento 87. La química francesa Marguerite Perey lo aisló entre los productos de desintegración del uranio. Se encontraba en cantidades muy pequeñas, y sólo los avances técnicos permitieron encontrarlo donde antes había pasado inadvertido. Dio al nuevo elemento el nombre de «francio», en honor de su país natal.
El elemento 85, al igual que el tecnecio, fue producido en el ciclotrón bombardeando bismuto (elemento 83). En 1940, Segré, Dale Raymond Corson y K. R. MacKenzie aislaron el elemento 85 en la Universidad de California, ya que Segré había emigrado de Italia a Estados Unidos. La Segunda Guerra Mundial interrumpió su trabajo sobre este elemento; pero, una vez acabada la contienda, el equipo reanudó su labor, y, en 1947, propuso para el elemento el nombre de «astato» (de la palabra griega que significa «inestable»). (Para entonces se habían encontrado en la Naturaleza pequeños restos de astato, como en el caso del francio, entre los productos de desintegración del uranio.)
Mientras tanto, el cuarto y último elemento de los que faltaban por descubrir (el 61) se había hallado entre los productos de fisión del uranio, proceso que explicamos en el capítulo 10. (También el tecnecio se encontró entre estos productos.) En 1945, tres químicos del Oak Ridge National Laboratory —J. A. Marinsky, L. E. Glendenin y Charles Dubois Coryell— aislaron el elemento 61. Lo denominaron «promecio» (promethium, voz inspirada en el nombre del dios Prometeo, que había robado su fuego al Sol para entregarlo a la Humanidad). Después de todo, el elemento 61 había sido «robado» a partir de los fuegos casi solares del horno atómico.
De este modo se completó la lista de los elementos, del 1 al 92. Sin embargo, en cierto sentido, la parte más extraña de la aventura acababa sólo de empezar. Porque los científicos habían rebasado los límites de la tabla periódica; el uranio no era el fin.
Elementos transuránidos
Ya en 1934 había empezado la búsqueda de los elementos situados más allá del uranio, o sea, los elementos «transuránidos». En Italia, Enrico Fermi comprobó que cuando bombardeaba un elemento con una partícula subatómica, recientemente descubierta, llamada «neutrón» (véase capítulo 7), ésta transformaba a menudo el elemento en el de número atómico superior más próximo. ¿Era posible que el uranio se transformase en el elemento 93, completamente sintético, que no existía en la Naturaleza? El equipo de Fermi procedió a bombardear el uranio con neutrones y obtuvo un producto que, al parecer, era realmente el elemento 93. Se le dio el nombre de «uranio X».
En 1938, Fermi recibió el premio Nobel de Física por sus estudios sobre el bombardeo con neutrones. Por aquella fecha, ni siquiera podía sospecharse la naturaleza real de su descubrimiento, ni sus consecuencias para la Humanidad. Al igual que Cristóbal Colón, había encontrado, no lo que estaba buscando, sino algo mucho más valioso, pero de cuya importancia no podía percatarse.
Basta decir, por ahora, que, tras seguir una serie de pistas que no condujeron a ninguna parte, descubrióse, al fin, que lo que Fermi había conseguido no era la creación de un nuevo elemento, sino la escisión del átomo de uranio en dos partes casi iguales. Cuando, en 1940, los físicos abordaron de nuevo el estudio de este proceso, el elemento 93 surgió como un resultado casi fortuito de sus experimentos. En la mezcla de elementos que determinaba el bombardeo del uranio por medio de neutrones, aparecía uno que, de principio, resistió todo intento de identificación. Entonces, Edwin Mc-Millan, de la Universidad de California, sugirió que quizá los neutrones liberados por fisión hubiesen convertido algunos de los átomos de uranio en un elemento de número atómico más alto, como Fermi había esperado que ocurriese. McMillan y Philip Abelson, un fisicoquímico, probaron que el elemento no identificado era, en realidad, el número 93. La prueba de su existencia la daba la naturaleza de su radiactividad, lo mismo que ocurriría en todos los descubrimientos subsiguientes.
McMillan sospechaba que pudiera estar mezclado con el número 93 otro elemento transuránido. El químico Glenn Theodore Seaborg y sus colaboradores Arthur Charles Wahl y J. W. Kennedy no tardaron en demostrar que McMillan tenía razón y que dicho elemento era el número 94.
De la misma forma que el uranio —elemento que se suponía el último de la tabla periódica— tomó su nombre de Urano, el planeta recientemente descubierto a la razón, los elementos 93 y 94 fueron bautizados, respectivamente, como «neptuno» y «plutonio», por Neptuno y Plutón, planetas descubiertos después de Urano. Y resultó que existía en la Naturaleza, pues más tarde se encontraron indicios de los mismos en menas de uranio. Así, pues, el uranio no era el elemento natural de mayor peso atómico.
Seaborg y un grupo de investigadores de la Universidad de California —entre los cuales destacaba Albert Ghiorso— siguieron obteniendo, uno tras otro, nuevos elementos transuránidos. Bombardeando plutonio con partículas subatómicas, crearon, en 1944, los elementos 95 y 96, que recibieron, respectivamente, los nombres de «americio» (por América) y «curio» (en honor de los Curie). Una vez obtenida una cantidad suficiente de americio y curio, bombardearon estos elementos y lograron obtener, en 1949, el número 97, y, en 1950, el 98. Estos nuevos elementos fueron llamados «berkelio» y «californio» (por Berkeley y California). En 1951, Seaborg y McMillan compartieron el premio Nobel de Química por esta serie de descubrimientos.
El descubrimiento de los siguientes elementos fue el resultado de unas investigaciones y pruebas menos pacíficas. Los elementos 99 y 100 surgieron en la primera explosión de una bomba de hidrógeno, la cual se llevó a cabo en el Pacífico, en noviembre de 1952. Aunque la existencia de ambos fue detectada en los restos de la explosión, no se confirmó ni se les dio nombres hasta después de que el grupo de investigadores de la Universidad de California obtuvo en su laboratorio, en 1955, pequeñas cantidades de ambos. Fueron denominados, respectivamente, «einstenio» y «fermio», en honor de Albert Einstein y Enrico Fermi, ambos, muertos unos meses antes. Después, los investigadores bombardearon una pequeña cantidad de «einstenio» y obtuvieron el elemento 101, al que denominaron «mendelevio», por Mendeléiev.
El paso siguiente llegó a través de la colaboración entre California y el Instituto Nobel de Suecia. Dicho instituto llevó a cabo un tipo muy complicado de bombardeo que produjo, aparentemente, una pequeña cantidad del elemento 102. Fue llamado «nobelio», en honor del Instituto; pero el experimento no ha sido confirmado. Se había obtenido con métodos distintos de los descritos por el primer grupo de investigadores. Mas, pese a que el «nobelio» no ha sido oficialmente aceptado como el nombre del elemento, no se ha propuesto ninguna otra denominación.
En 1961 se detectaron algunos átomos del elemento 103 en la Universidad de California, a los cuales se les dio el nombre de «laurencio» (por E. O. Lawrence, que había fallecido recientemente). En 1964, un grupo de científicos soviéticos, bajo la dirección de Georguéi Nikoláievich Flerov, informó sobre la obtención del elemento 104, y en 1965, sobre la del 105. En ambos casos, los métodos usados para formar los elementos no pudieron ser confirmados. El equipo americano dirigido por Albert Ghioso obtuvo también dichos elementos, independientemente de los soviéticos. Entonces se planteó la discusión acerca de la prioridad; ambos grupos reclamaban el derecho a dar nombre a los nuevos elementos. El grupo soviético llamó al elemento 104 «kurchatovio», en honor de Igor Vasilievich Kurchatov, el cual había dirigido al equipo soviético que desarrolló la bomba atómica rusa, y que murió en 1960. Por su parte, el grupo americano dio al elemento 104 el nombre de «rutherfordio», y al 105 el de «hahnio», en honor, respectivamente, de Ernest Rutherford y Otto Hahn, los cuales dieron las claves para los descubrimientos de la estructura subatómica.
Elementos superpesados
Cada paso en esta ascensión de la escala transuránida fue más difícil que el anterior. En cada estadio sucesivo, el elemento se hizo más difícil de acumular y más inestable. Cuando se llegó al mendelevio, la identificación tuvo que hacerse sobre la base de diecisiete átomos, y no más. Afortunadamente, las técnicas de detección de la radiación se mejoraron maravillosamente en 1955. Los científicos de Berkeley conectaban sus instrumentos a un avisador, con lo que, cada vez que se formaba un átomo de mendelevio, la radiación característica emitida quedaba anunciada por un grave y triunfante avisador de incendios. (De todos modos, el Departamento de extinción de incendios lo prohibió pronto…)
Los elementos superiores fueron detectados incluso en las condiciones más rarificadas. Un solo átomo de un elemento deseado puede detectarse al observar en detalle los productos de su desintegración.
¿Existe necesidad de tratar de llegar más lejos, más allá del 105, aparte del escalofrío propio de batir un récord y de dar el nombre de uno en el libro correspondiente como descubridor de un elemento? (Lavoisier, el mayor de todos los químicos, nunca consiguió ningún descubrimiento, y su fracaso le preocupó en extremo.)
Aún queda por hacer un importante y posible descubrimiento. El incremento en inestabilidad a medida que se asciende en la escala de los números atómicos es uniforme. El más complejo de los átomos estables es el bismuto (83). Detrás del mismo, los seis elementos del 84 al 89 inclusive son tan inestables que cualquier cantidad presente en el momento de la formación de la Tierra ya habría desaparecido en la actualidad. Y luego, y más bien sorprendentemente, sigue el torio (90) y el uranio (92), que son casi estables. Del torio y el uranio existentes en la Tierra en el momento de su formación, el 80 % del primero y el 50 % del último existen aún hoy. Los físicos han elaborado teorías de la estructura atómica para tener esto en cuenta (como explicaré en el capítulo siguiente); y si esas teorías son correctas, en ese caso los elementos 110 y 114 deberían ser más estables de lo que se esperaría de ellos dados sus elevados números atómicos. Por lo tanto, existe considerable interés en conseguir esos elementos, como una forma de comprobar las teorías.
En 1976, se produjo un informe de ciertos halos (marcas circulares negras en la mica) que indicarían la presencia de esos elementos superpesados. Los halos surgen de la radiación emitida por pequeñas cantidades de torio y uranio, pero existen unos halos un poco más allá de lo normal que deben surgir de unos átomos más energéticamente radiactivos que, sin embargo, son lo suficientemente estables como para haber persistido hasta los tiempos modernos. Y debía de tratarse de los superpesados. Por desgracia, las deducciones no se vieron apoyadas en general por los científicos, y dicha sugerencia fue olvidada. Los científicos siguen buscando.
Electrones
Cuando Mendeléiev y sus contemporáneos descubrieron que podían distribuir los elementos en una tabla periódica compuesta por familias de sustancias de propiedades similares, no tenían noción alguna acerca del porqué los elementos pertenecían a tales grupos o del motivo por el que estaban relacionadas las propiedades. De pronto surgió una respuesta simple y clara, aunque tras una larga serie de descubrimientos, que al principio no parecían tener relación con la Química.
Todo empezó con unos estudios sobre la electricidad. Faraday realizó con la electricidad todos los experimentos imaginables; incluso trató de enviar una descarga eléctrica a través del vacío. Mas no pudo conseguir un vacío lo suficientemente perfecto para su propósito. Pero en 1854, un soplador de vidrio alemán, Heinrich Geissler, inventó una bomba de vacío adecuada y fabricó un tubo de vidrio en cuyo interior iban electrodos de metal en un vacío de calidad sin precedentes hasta entonces. Cuando se logró producir descargas eléctricas en el «tubo de Geissler», comprobóse que en la pared opuesta al electrodo negativo aparecía un resplandor verde. El físico alemán Eugen Goldstein sugirió, en 1876, que tal resplandor verde se debía al impacto causado en el vidrio por algún tipo de radiación originada en el electrodo negativo, que Faraday había denominado «cátodo». Goldstein dio a la radiación el nombre de «rayos catódicos».
¿Eran los rayos catódicos una forma de radiación electromagnética? Goldstein lo creyó así; en cambio, lo negaron el físico inglés William Crookes y algunos otros, según los cuales, dichos rayos eran una corriente de partículas de algún tipo. Crookes diseñó versiones mejoradas del tubo de Geissler (llamadas «tubos Crookes»), con las cuales pudo demostrar que los rayos eran desviados por un imán. Esto quizá significaba que dichos rayos estaban formados por partículas cargadas eléctricamente.
En 1897, el físico Joseph John Thomson zanjó definitivamente la cuestión al demostrar que los rayos catódicos podían ser también desviados por cargas eléctricas. ¿Qué eran, pues, las «partículas» catódicas? En aquel tiempo, las únicas partículas cargadas negativamente que se conocían eran los iones negativos de los átomos. Los experimentos demostraron que las partículas de los rayos catódicos no podían identificarse con tales iones, pues al ser desviadas de aquella forma por un campo electromagnético, debían de poseer una carga eléctrica inimaginablemente elevada, o bien tratarse de partículas muy ligeras, con una masa mil veces más pequeña que la de un átomo de hidrógeno. Esta última interpretación era la que encajaba mejor en el marco de las pruebas realizadas. Los físicos habían ya intuido que la corriente eléctrica era transportada por partículas. En consecuencia, estas partículas de rayos catódicos fueron aceptadas como las partículas elementales de la electricidad. Se les dio el nombre de «electrones», denominación sugerida, en 1891, por el físico irlandés George Johnstone Stoney. Finalmente, se determinó que la masa del electrón era 1.837 veces menor que la de un átomo de hidrógeno. (En 1906, Thomson fue galardonado con el premio Nobel de Física por haber establecido la existencia del electrón.)
El descubrimiento del electrón sugirió inmediatamente que debía de tratarse de una subpartícula del átomo. En otras palabras, que los átomos no eran las unidades últimas indivisibles de la materia que habían descrito Demócrito y John Dalton.
Aunque costaba trabajo creerlo, las pruebas convergían de manera inexorable. Uno de los datos más convincentes fue la demostración, hecha por Thomson, de que las partículas con carga negativa emitidas por una placa metálica al ser incidida por radiaciones ultravioleta (el llamado «efecto fotoeléctrico»), eran idénticas a los electrones de los rayos catódicos. Los electrones fotoeléctricos debían de haber sido arrancados de los átomos del metal.
La periodicidad de la tabla periódica
Puesto que los electrones podían separarse fácilmente de los átomos, tanto por el efecto fotoeléctrico como por otros medios, era natural llegar a la conclusión de que se hallaban localizados en la parte exterior del átomo. De ser así, debía de existir una zona cargada positivamente en el interior del átomo, que contrarrestaría las cargas negativas de los electrones, puesto que el átomo, globalmente considerado, era neutro. En este momento, los investigadores empezaron a acercarse a la solución del misterio de la tabla periódica.
Separar un electrón de un átomo requiere una pequeña cantidad de energía. De acuerdo con el mismo principio, cuando un electrón ocupa un lugar vacío en el átomo, debe ceder una cantidad igual de energía. (La Naturaleza es generalmente simétrica, en especial cuando se trata de energía.) Esta energía es liberada en forma de radiación electromagnética. Ahora bien, puesto que la energía de la radiación se mide en términos de longitud de onda, la longitud de onda de la radiación emitida por un electrón que se une a un determinado átomo indicarán la fuerza con que el electrón es sujetado por este átomo. La energía de la radiación aumentaba al acortarse la longitud de onda: cuanto mayor es la energía, más corta es la longitud de onda.
Y con esto llegamos al descubrimiento, hecho por Moseley, de que los metales —es decir, los elementos más pesados— producen rayos X, cada uno de ellos con su longitud de onda característica, que disminuye de forma regular, a medida que se va ascendiendo en la tabla periódica. Al parecer, cada elemento sucesivo retenía sus electrones con más fuerza que el anterior, lo cual no es más que otra forma de decir que cada uno de ellos tiene una carga positiva más fuerte, en su región interna, que el anterior.
Suponiendo que, en un electrón, a cada unidad de carga positiva le corresponde una de carga negativa, se deduce que el átomo de cada elemento sucesivo de la tabla periódica debe tener un electrón más. Entonces, la forma más simple de formar la tabla periódica consiste en suponer que el primer elemento, el hidrógeno, tiene una unidad de carga positiva y un electrón; el segundo elemento, el helio, 2 cargas positivas y 2 electrones; el tercero, el litio, 3 cargas positivas y 3 electrones, y así, hasta llegar al uranio, con 92 electrones. De este modo, los números atómicos de los elementos han resultado ser el número de electrones de sus átomos.
Una prueba más, y los científicos atómicos tendrían la respuesta a la periodicidad de la tabla periódica. Se puso de manifiesto que la radiación de electrones de un determinado elemento no estaba necesariamente restringida a una longitud de onda única; podía emitir radiaciones de dos, tres, cuatro e incluso más longitudes de onda distintas. Estas series de radiaciones fueron denominadas K, L, M, etc. Los investigadores interpretaron esto como una prueba de que los electrones estaban dispuestos en «capas» alrededor del núcleo del átomo de carga positiva. Los electrones de la capa más interna eran sujetados con mayor fuerza, y para conseguir su separación se necesitaba la máxima energía, es decir, de longitudes de onda más corta, o de la serie K. Los electrones de la capa siguiente emitían la serie L de radiaciones; la siguiente capa producía la serie M, etc. En consecuencia, estas capas fueron denominadas K, L, M, etc.
Hacia 1925, el físico austríaco Wolfgang Pauli enunció su «principio de exclusión», el cual explicaba la forma en que los electrones estaban distribuidos en el interior de cada capa, puesto que, según este principio, dos electrones no podían poseer exactamente la misma energía ni el mismo spin. Por este descubrimiento, Pauli recibió el premio Nobel de Física en 1945.
Los gases nobles o inertes
En 1916, el químico americano Gilbert-Newton Lewis determinó las similitudes de las propiedades y el comportamiento químico de algunos de los elementos más simples sobre la base de su estructura en capas. Para empezar, había pruebas suficientes de que la capa más interna estaba limitada a dos electrones. El hidrógeno sólo tiene un electrón; por tanto, la capa está incompleta. El átomo tiende a completar esta capa K, y puede hacerlo de distintas formas. Por ejemplo, dos átomos de hidrógeno pueden compartir sus respectivos electrones y completar así mutuamente sus capas K. Ésta es la razón de que el hidrógeno se presente casi siempre en forma de un par de átomos: la molécula de hidrógeno. Se necesita una gran cantidad de energía para separar los dos átomos y liberarlos en forma de «hidrógeno atómico». Irving Langmuir, de la «General Electric Company» —quien, independientemente, llegó a un esquema similar, que implicaba los electrones y el comportamiento químico— llevó a cabo una demostración práctica de la intensa tendencia del átomo de hidrógeno a mantener completa su capa de electrones. Obtuvo una «antorcha de hidrógeno atómico» soplando gas de hidrógeno a través de un arco eléctrico, que separaba los átomos de las moléculas; cuando los átomos se recombinaban, tras pasar el arco, liberaban las energías que habían absorbido al separarse, lo cual bastaba para alcanzar temperaturas superiores a los 3.400 °C.
En el helio (elemento 2), la capa K está formada por dos electrones. Por tanto, los átomos de helio son estables y no se combinan con otros átomos. Al llegar al litio (elemento 3), vemos que dos de sus electrones completan la capa K y que el tercero empieza la capa L. Los elementos siguientes añaden electrones a esta capa, uno a uno: el berilio tiene 2 electrones en la capa L; el boro, 3; el carbono, 4; el nitrógeno, 5; el oxígeno, 6; el flúor, 7, y el neón 8. Ocho es el límite para la capa L, por lo cual el neón, lo mismo que el helio, tiene su capa exterior de electrones completa. Y, desde luego, es también un gas inerte, con propiedades similares a las del helio.
Cada átomo cuya capa exterior no está completa, tiende a combinarse con otros átomos, de forma que pueda completarla. Por ejemplo, el átomo de litio cede fácilmente su único electrón en la capa L, de modo que su capa exterior sea la K, completa, mientras que el flúor tiende a captar un electrón, que añade a los siete que ya tiene, para completar su capa L. Por tanto, el litio y el flúor tienen afinidad el uno por el otro; y cuando se combinan, el litio cede su electrón L al flúor, para completar la capa L exterior de este último. Dado que no cambia las cargas positivas del interior del átomo, el litio, con un electrón de menos, es ahora portador de una carga positiva, mientras que el flúor, con un electrón de más, lleva una carga negativa. La mutua atracción de las cargas opuestas mantiene unidos a los dos iones. El compuesto se llama fluoruro de litio.
Los electrones de la capa L pueden ser compartidos o cedidos. Por ejemplo, uno de cada dos átomos de flúor puede compartir uno de sus electrones con el otro, de modo que cada átomo tenga un total de ocho en su capa L, contando los dos electrones compartidos. De forma similar, dos átomos de oxígeno compartirán un total de cuatro electrones para completar sus capas L; y dos átomos de nitrógeno compartirán un total de 6. De este modo, el flúor, el oxígeno y el nitrógeno forman moléculas de dos átomos.
El átomo de carbono, con sólo cuatro electrones en su capa L, compartirá cada uno de ellos con un átomo distinto de hidrógeno, para completar así las capas K de los cuatro átomos de hidrógeno. A su vez, completa su propia capa L al compartir sus electrones. Esta disposición estable es la molécula de metano CH4.
Del mismo modo, un átomo de nitrógeno compartirá los electrones con tres átomos de hidrógeno para formar el amoníaco; un átomo de oxígeno compartirá sus electrones con dos átomos de hidrógeno para formar el agua; un átomo de carbono compartirá sus electrones con dos átomos de oxígeno para formar anhídrido carbónico; etc. Casi todos los compuestos formados por elementos de la primera parte de la tabla periódica pueden ser clasificados de acuerdo con esta tendencia a completar su capa exterior cediendo electrones, aceptando o compartiendo electrones.
El elemento situado después del neón, el sodio, tiene 11 electrones, y el undécimo debe empezar una tercera capa. Luego sigue el magnesio, con 2 electrones en la capa M; el aluminio, con 3; el silicio, con 4; el fósforo, con 5; el azufre, con 6; el cloro, con 7, y el argón, con 8.
Ahora bien, cada elemento de este grupo corresponde a otro de la serie anterior. El argón, con 8 electrones en la capa M, se asemeja al neón (con 8 electrones en la capa L) y es un gas inerte. El cloro, con 7 electrones en su capa exterior, se parece mucho al flúor en sus propiedades químicas. Del mismo modo, el silicio se parece al carbono; el sodio, al litio, etc. (fig. 6.2).

Fig. 6.2. Cesión y compartimento de electrones. El litio cede el electrón de su capa exterior al flúor, en la combinación fluoruro de litio; cada átono tiene entonces una capa eterna completa. En la molécula de flúor (F2), se comparten dos electrones, que completan las capas exteriores de ambos átomos.
Así ocurre a lo largo de toda la tabla periódica. Puesto que el comportamiento químico de cada elemento depende de la configuración de los electrones de su capa exterior, todos los que, por ejemplo, tengan un electrón en la capa exterior, reaccionarán químicamente de un modo muy parecido. Así, todos los elementos de la primera columna de la tabla periódica —litio, sodio, potasio, rubidio, cesio e incluso el francio, el elemento radiactivo hecho por el hombre— son extraordinariamente parecidos en sus propiedades químicas. El litio tiene 1 electrón en la capa L; el sodio, 1 en la M; el potasio, 1 en la N; el rubidio, 1 en la O; el cesio, 1 en la P, y el francio, 1 en la Q. Una vez más, se parecen entre sí todos los elementos con siete electrones en sus respectivas capas exteriores (flúor, cloro, bromo, yodo y astato). Lo mismo ocurre con la última columna de la tabla, el grupo, de capa completa, que incluye el helio, neón, argón, criptón, xenón y radón.
El principio de Lewis-Langmuir se cumple de forma tan perfecta, que sirve aún, en su forma original, para explicar las variedades de comportamiento más simples y directas entre los elementos. Sin embargo, no todos los comportamientos son tan simples ni tan directos como pueda creerse.
Por ejemplo, cada uno de los gases inertes —helio, neón, argón, criptón, xenón y radón— tiene ocho electrones en la capa exterior (a excepción del helio, que tiene dos en su única capa), situación que es la más estable posible. Los átomos de estos elementos tienen una tendencia mínima a perder o ganar electrones, y, por tanto, a tomar parte en reacciones químicas. Estos gases, tal como indica su nombre, serían «inertes».
Sin embargo, una «tendencia mínima» no es lo mismo que «sin tendencia alguna»; pero la mayor parte de los químicos lo olvidó, y actuó como si fuese realmente imposible para los gases inertes formar compuestos. Por supuesto que ello no ocurría así con todos. Ya en 1932, el químico americano Linus Pauling estudió la facilidad con que los electrones podían separarse de los distintos elementos, y observó que todos los elementos sin excepción, incluso los gases inertes, podían ser desprovistos de electrones. La única diferencia estribaba en que, para que ocurriese esto, se necesitaba más energía en el caso de los gases inertes que en el de los demás elementos situados junto a ellos en la tabla periódica.
La cantidad de energía requerida para separar los electrones en los elementos de una determinada familia, disminuye al aumentar el peso atómico, y los gases inertes más pesados, el xenón y el radón, no necesitan cantidades excesivamente elevadas. Por ejemplo, no es más difícil extraer un electrón a partir de un átomo de xenón que de un átomo de oxígeno.
Por tanto, Pauling predijo que los gases inertes más pesados podían formar compuestos químicos con elementos que fueran particularmente propensos a aceptar electrones. El elemento que más tiende a aceptar electrones es el flúor, y éste parecía ser el que naturalmente debía elegirse.
Ahora bien, el radón, el gas inerte más pesado, es radiactivo y sólo puede obtenerse en pequeñísimas cantidades. Sin embargo, el xenón, el siguiente gas más pesado, es estable y se encuentra en pequeñas cantidades en la atmósfera. Por tanto, lo mejor sería intentar formar un compuesto entre el xenón y el flúor. Sin embargo, durante 30 años no se pudo hacer nada a este respecto, principalmente porque el xenón era caro, y el flúor, muy difícil de manejar, y los químicos creyeron que era mejor dedicarse a cosas menos complicadas.
No obstante, en 1962, el químico anglocanadiense Neil Bartlett, trabajando con un nuevo compuesto, el hexafluoruro de platino (F6Pt), manifestó que se mostraba notablemente ávido de electrones, casi tanto como el propio flúor. Este compuesto tomaba electrones a partir del oxígeno, elemento que tiende más a ganar electrones que a perderlos. Si el F6Pt podía captar electrones a partir del oxígeno, debía de ser capaz también de captarlos a partir del xenón. Se intentó el experimento, y se obtuvo el fluoroplatinato de xenón (F6PtXe), primer compuesto de un gas inerte.
Otros químicos se lanzaron en seguida a este campo de investigación, y se obtuvo cierto número de compuestos de xenón con flúor, con oxígeno o con ambos, el más estable de los cuales fue el difluoruro de xenón (F2Xe). Formóse asimismo un compuesto de criptón y flúor: el tetrafluoruro de criptón (F4Kr), así como otros de radón y flúor. También se formaron compuestos con oxígeno. Había, por ejemplo, oxitetrafluoruro de xenón (OF4Xe), ácido xénico (H2O4Xe) y perxenato de sodio (XeO6Na4), que explota fácilmente y es peligroso. Los gases inertes más livianos — argón, neón y helio— ofrecen mayor resistencia a compartir sus electrones que los más pesados, por lo cual permanecen inertes (según las posibilidades actuales de los químicos).
Los químicos no tardaron en recuperarse del shock inicial que supuso descubrir que los gases inertes podían formar compuestos. Después de todo, tales compuestos encajaban en el cuadro general. En consecuencia, hoy existe una aversión general a denominar «gases inertes» a estos elementos. Se prefiere el nombre de «gases nobles», y se habla de «compuestos de gases nobles» y «Química de los gases nobles». (Creo que se trata de un cambio para empeorar. Al fin y al cabo, los gases siguen siendo inertes, aunque no del todo. En este contexto, el concepto «noble» implica «reservado» o «poco inclinado a mezclarse con la manada», lo cual resulta tan inapropiado como «inerte» y, sobre todo, no anda muy de acuerdo con una «sociedad democrática».)
Los elementos tierras raras
El esquema de Lewis-Langmuir que se aplicó demasiado rígidamente a los gases inertes, apenas puede emplearse para muchos de los elementos cuyo número atómico sea superior a 20. En particular se necesitaron ciertos perfeccionamientos para abordar un aspecto muy sorprendente de la tabla periódica, relacionado con las llamadas «tierras raras» (los elementos 57 al 71, ambos inclusive).
Retrocediendo un poco en el tiempo, vemos que los primeros químicos consideraban como «tierra» —herencia de la visión griega de la «tierra» como elemento— toda sustancia insoluble en agua y que no pudiera ser transformada por el calor. Estas sustancias incluían lo que hoy llamaríamos óxido de calcio, óxido de magnesio, bióxido silícico, óxido férrico, óxido de aluminio, etc., compuestos que actualmente constituyen alrededor de un 90 % de la corteza terrestre. Los óxidos de calcio y magnesio son ligeramente solubles, y en solución muestran propiedades «alcalinas» (es decir, opuestas a las de los ácidos), por lo cual fueron denominados «tierras alcalinas»; cuando Humphry Davy aisló los metales calcio y magnesio partiendo de estas tierras, se les dio el nombre de metales alcalinotérreos. De la misma forma se designaron eventualmente todos los elementos que caben en la columna de la tabla periódica en la que figuran el magnesio y el calcio; es decir, el berilio, estroncio, bario y radio.
El rompecabezas empezó en 1794, cuando un químico finlandés, Johan Gadolin, examinó una extraña roca que había encontrado cerca de la aldea sueca de Ytterby, y llegó a la conclusión de que se trataba de una nueva «tierra». Gadolin dio a esta «tierra rara» el nombre de «itrio» (por Ytterby). Más tarde, el químico alemán Martin Heinrich Klaproth descubrió que el itrio podía dividirse en dos «tierras», para una de las cuales siguió conservando el nombre de itrio, mientras que llamó a la otra «cerio» (por el planetoide Ceres, recientemente descubierto). Pero, a su vez, el químico sueco Cari Gustav Mosander separó éstos en una serie de tierras distintas. Todas resultaron ser óxidos de nuevos elementos, denominados «metales de las tierras raras». En 1907 se habían identificado ya 14 de estos elementos. Por orden creciente de peso atómico son:
lantano (voz tomada de la palabra griega que significa «escondido»)
cerio (de Ceres)
praseodimio (del griego «gemelo verde», por la línea verde que da su espectro)
neodimio («nuevos gemelos»)
samarío (de «samarsquita», el mineral en que se encontró)
europio (de Europa)
gadolinio (en honor de Johan Gadolin)
terbio (de Ytterby)
disprosio (del griego «difícil de llegar a»)
holmio (de Estocolmo)
erbio (de Ytterby)
tulio (de Thule, antiguo nombre de Escandinavia) iterbio (de Ytterby)
lutecio (de Lutecia, nombre latino de París).
Basándose en sus propiedades de rayos X, estos elementos recibieron los números atómicos 57 (lantano) a 71 (lutecio). Como ya hemos dicho, existía un vacío en el espacio 61 hasta que el elemento incógnito, el promecio, emergió a partir de la fisión del uranio. Era el número 15 de la lista.
Ahora bien, el problema planteado por los elemento de las tierras raras es el de que, aparentemente, no encajan en la tabla periódica. Por suerte, sólo se conocían cuatro cuando Mendeléiev propuso la tabla; si se hubiesen conocido todos, la tabla podría haber resultado demasiado confusa para ser aceptada. Hay veces, incluso en la Ciencia, en que la ignorancia es una suerte.
El primero de los metales de las tierras raras, el lantano, encaja perfectamente con el itrio, número 39, el elemento situado por encima de él en la tabla. (El itrio, aunque fue encontrado en las mismas menas que las tierras raras y es similar a ellas en sus propiedades, no es un metal de tierra rara. Sin embargo, toma también su nombre de la aldea sueca de Ytterby. Así, cuatro elementos se denominan partiendo del mismo origen, lo cual parece excesivo.) La confusión empieza con las tierras raras colocadas después del lantano, principalmente el cerio, que debería parecerse al elemento que sigue al itrio, o sea, al circonio. Pero no es así, pues se parece al itrio. Lo mismo ocurre con los otros quince elementos de las tierras raras; se parecen mucho al itrio y entre sí (de hecho, son tan químicamente parecidos, que al principio pudieron separarse sólo por medio de procedimientos muy laboriosos), pero no están relacionados con ninguno de los elementos que les preceden en la tabla. Prescindamos del grupo de tierras raras y pasemos al hafnio, el elemento 72, en el cual encontraremos el elemento relacionado con el circonio, colocado después del itrio.

Fig. 6.3. Las capas de electrones del lantano. Nótese que la cuarta subcapa de la capa N ha sido omitida y está vacía.
Desconcertados por este estado de cosas, lo único que pudieron hacer los químicos fue agrupar todos los elementos de tierras raras en un espacio situado debajo del itrio, y alineados uno por uno, en una especie de nota al pie de la tabla.
Los elementos transicionales
Finalmente, la respuesta a este rompecabezas llegó como resultado de detalles añadidos al esquema de Lewis-Langmuir sobre la estructura de las capas de electrones en los elementos.
En 1921, C. R. Bury sugirió que el número de electrones de cada capa no estaba limitado necesariamente a ocho. El ocho era el número que bastaba siempre para satisfacer la capacidad de la capa exterior. Pero una capa podía tener un mayor número de electrones si no estaba en el exterior. Como quiera que las capas se iban formando sucesivamente, las más internas podían absorber más electrones, y cada una de las siguientes podía retener más que la anterior. Así, la capacidad total de la capa K sería de 2 electrones; la de la L, de 8; la de la M, de 18; la de la N, de 32, y así sucesivamente. Este escalonamiento se ajusta al de una serie de sucesivos cuadrados multiplicados por 2 (por ejemplo, 2·1, 2·4, 2·9, 2·16, etc.).
Este punto de vista fue confirmado por un detenido estudio del espectro de los elementos. El físico danés Niels Henrik David Bohr demostró que cada capa de electrones estaba constituida por subcapas de niveles de energía ligeramente distintos. En cada capa sucesiva, las subcapas se hallan más separadas entre sí, de tal modo que pronto se imbrican las capas. En consecuencia, la subcapa más externa de una capa interior (por ejemplo, la M), puede estar realmente más lejos del centro que la subcapa más interna de la capa situada después de ella (por ejemplo, la N). Por tanto, la subcapa interna de la capa N puede estar llena de electrones, mientras que la subcapa exterior de la capa M puede hallarse aún vacía.
Un ejemplo aclarará esto. Según esta teoría, la capa M está dividida en tres subcapas, cuyas capacidades son de 2, 6 y 10 electrones, respectivamente, lo cual da un total de 18. El argón, con 8 electrones en su capa M, ha completado sólo 2 subcapas internas. Y, de hecho, la tercera subcapa, o más externa, de la capa M, no conseguirá el próximo electrón en el proceso de formación de elementos, al hallarse por debajo de la subcapa más interna de la capa N. Así, en el potasio —elemento que sigue al argón—, el electrón decimonoveno no se sitúa en la subcapa más exterior de M, sino en la subcapa más interna de N. El potasio, con un electrón en su capa N, se parece al sodio, que tiene un electrón en su capa M. El calcio —el siguiente elemento (20)— tiene dos electrones en la capa N y se parece al magnesio, que posee dos en la capa M. Pero la subcapa más interna de la capa N, que tiene capacidad sólo para 2 electrones, está completa. Los siguientes electrones que se han de añadir pueden empezar llenando la subcapa más exterior de la capa M, que hasta entonces ha permanecido inalterada. El escandio (21) inicia el proceso, y el cinc (30) lo termina. En el cinc, la subcapa más exterior de la capa M adquiere, por fin, los electrones que completan el número de 10. Los 30 electrones del cinc están distribuidos del siguiente modo: 2 en la capa K,8 en la L,18 en la M y 2 en la N. Al llegar a este punto, los electrones pueden seguir llenando la capa N. El siguiente electrón constituye el tercero de la capa N y forma el galio (31), que se parece al aluminio, con 3 electrones en la capa M.
Lo más importante de este proceso es que los elementos 21 al 30 —los cuales adquieren una configuración parecida para completar una subcapa que había sido omitida temporalmente— son «de transición». Nótese que el calcio se parece al magnesio, y el galio, al aluminio. El magnesio y el aluminio están situados uno junto a otro en la tabla periódica (números 12 y 13). En cambio, no lo están el calcio (20) ni el galio (31). Entre ellos se encuentran los elementos de transición, lo cual hace aún más compleja la tabla periódica.
La capa N es mayor que la M y está dividida en cuatro subcapas, en vez de tres: puede tener 2, 6, 10 y 14 electrones, respectivamente. El criptón (elemento 36) completa las dos subcapas más internas de la capa N; pero aquí interviene la subcapa más interna de la capa O, que está superpuesta, y antes de que los electrones se sitúen en las dos subcapas más externas de la N, deben llenar dicha subcapa. El elemento que sigue al criptón, el rubidio (37), tiene su electrón número 37 en la capa O. El estroncio (38) completa la subcapa O con dos electrones. De aquí en adelante, nuevas series de elementos de transición rellenan la antes omitida tercera subcapa de la capa N. Este proceso se completa en el cadmio (48); se omite la subcapa cuarta y más exterior de N, mientras los electrones pasan a ocupar la segunda subcapa interna de O, proceso que finaliza en el xenón (54).
Pero ahora, a nivel de la cuarta subcapa de N, es tan manifiesta la superposición, que incluso la capa 9 interpone una subcapa, la cual debe ser completada antes que la última de N. Tras el xenón viene el cesio (55) y el bario (56), con uno y dos electrones, respectivamente, en la capa P. Aún no ha llegado el turno a N: el electrón 57 va a parar a la tercera subcapa de la capa O, para crear el lantano. Entonces, y sólo entonces, entra, por fin, un electrón en la subcapa más exterior de la capa N. Uno tras otro, los elementos de tierras raras añaden electrones a la capa H, hasta llegar al elemento 71 (el lutecio), que la completa. Los electrones del lutecio están dispuestos del siguiente modo: 2 en la capa K, 8 en la L, 18 en la M, 32 en la N, 9 en la O (dos subcapas llenas, más un electrón en la subcapa siguiente) y 2 en la P (cuya subcapa más interna está completa) (fig. 6.4).

Fig. 6.4. La representación esquemática de la imbricación de capas y subcapas electrónicas en el lantano. La subcapa más exterior de la capa N aún no ha sido completada.
Finalmente, empezamos a comprender por qué son tan parecidos los elementos de tierras raras y algunos otros grupos de elementos de transición. El factor decisivo que diferencia a los elementos, por lo que respecta a sus propiedades químicas, es la configuración de electrones en su capa más externa. Por ejemplo, el carbono, con 4 electrones en su capa exterior, y el nitrógeno, con 5, son completamente distintos en sus propiedades. Por otra parte, las propiedades varían menos en las secuencias de elementos en que los electrones están destinados a completar sus subcapas más internas, mientras la capa más externa permanece inalterable. Así, son muy parecidos en su comportamiento químico el hierro, el cobalto y el níquel (elementos 26, 27 y 28), todos los cuales tienen la misma configuración electrónica en la capa más externa, una subcapa N llena con dos electrones. Sus diferencias en la configuración electrónica interna (en una subcapa M) están enmascaradas en gran parte por su similitud electrónica superficial. Y esto es más evidente aún en los elementos de tierras raras. Sus diferencias (en la capa N) quedan enterradas no bajo una, sino bajo dos configuraciones electrónicas externas (en las capas O y P), que en todos estos elementos son idénticas. Constituye una pequeña maravilla el hecho de que los elementos sean químicamente tan iguales como los guisantes en su vaina.
Como quiera que los metales de tierras raras tienen tan pocos usos y son tan difíciles de separar, los químicos hicieron muy pocos esfuerzos para conseguirlo, hasta que se logró fisionar el átomo de uranio. Luego, el separarlos se convirtió en una tarea muy urgente, debido a que las variedades radiactivas de alguno de estos elementos se encontraban entre los principales productos de la fisión, y en el proyecto de la bomba atómica era necesario separarlos e identificarlos rápida y claramente.
El problema fue resuelto en breve plazo con ayuda de una técnica química creada, en 1906, por el botánico ruso Mijaíl Seménovich Tswett, quien la denominó «cromatografía» («escritura en color»). Tswett descubrió que podía separar pigmentos vegetales químicamente muy parecidos haciéndolos pasar, en sentido descendente, a través de una columna de piedra caliza en polvo, con ayuda de un disolvente. Tswett disolvió su mezcla de pigmentos vegetales en éter de petróleo y vertió esta mezcla sobre la piedra caliza. Luego incorporó disolvente puro. A medida que los pigmentos eran arrastrados por el líquido a través del polvo de piedra caliza, cada uno de ellos se movía a una velocidad distinta, porque su grado de adherencia al polvo era diferente. El resultado fue que se separaron en una serie de bandas, cada una de ellas de distinto color.
Al seguir lavando las sustancias separadas, iban apareciendo aisladas en el extremo inferior de la columna, de la que eran recogidas.
Durante muchos años, el mundo de la Ciencia ignoró el descubrimiento de Tswett, quizá porque se trataba sólo de un botánico y, además, ruso, cuando, a la sazón, eran bioquímicos alemanes las máximas figuras de la investigación sobre técnicas para separar sustancias difíciles de individualizar. Pero en 1931, un bioquímico, y precisamente alemán, Richard Willstátter, redescubrió el proceso, que entonces sí se generalizó. (Willstátter había recibido el premio Nobel de Química en 1915 por su excelente trabajo sobre pigmentos vegetales. Y, por lo que sabemos, Tswett no ha recibido honor alguno.)
La cromatografía a través de columnas de materiales pulverizados mostróse como un procedimiento eficiente para toda clase de mezclas, coloreadas o no. El óxido de aluminio y el almidón resultaron mejores que la piedra caliza para separar moléculas corrientes. Cuando se separan iones, el proceso se llama «intercambio de iones», y los compuestos conocidos con el nombre de zeolitas fueron los primeros materiales aplicados con este fin. Los iones de calcio y magnesio podrían ser extraídos del agua «dura», por ejemplo, vertiendo el agua a través de una columna de zeolita. Los iones de calcio y magnesio se adhieren a ella y son remplazados, en la solución, por iones de sodio que contiene la zeolita, de modo que al pie de la columna van apareciendo gotas de agua «blanda». Los iones de sodio de la zeolita deben ser remplazados de vez en cuando vertiendo en la columna una solución concentrada de sal corriente (cloruro sódico). En 1935 se perfeccionó el método al desarrollarse las «resinas intercambiadoras de iones», sustancias sintéticas que pueden ser creadas especialmente para el trabajo que se ha de realizar. Por ejemplo, ciertas resinas sustituyen los iones de hidrógeno por iones positivos, mientras que otras sustituyen iones hidroxilos por iones negativos. Una combinación de ambos tipos permitiría extraer la mayor parte de las sales del agua de mar. Cajitas que contenían esas resinas formaban parte de los equipos de supervivencia durante la Segunda Guerra Mundial.
El químico americano Frank Harold Spedding fue quien aplicó la cromatografía de intercambio de iones a la separación de las tierras raras. Descubrió que estos elementos salían de una columna de intercambio de iones en orden inverso a su número atómico, de modo que no sólo se separaban rápidamente, sino que también se identificaban. De hecho, el descubrimiento del promecio, el incógnito elemento 61, fue confirmado a partir de las pequeñas cantidades encontradas entre los productos de fisión.
Gracias a la cromatografía puede prepararse hasta 1 t de elementos de tierras raras purificados. Pero resulta que las tierras raras no son especialmente raras. En efecto, la más rara (a excepción del promecio) es más común que el oro o la plata, y las más corrientes —lantano, cerio y neodimio— abundan más que el plomo. En conjunto, los metales de tierras raras forman un porcentaje más importante de la corteza terrestre que el cobre y el estaño juntos. De aquí que los científicos sustituyeran el término «tierras raras» por el de «lantánidos», en atención al más importante de estos elementos. En realidad, los lantánidos individuales no han sido muy usados en el pasado, pero la finalidad de la separación actual ha multiplicado sus empleos y, hacia los años 1970, se usaban ya 12.000.000 de kilos. El mischmetal, una mezcla que consiste, principalmente, en cerio, lantano y neodimio, constituye las tres cuartas partes del peso de las piedras para mecheros de fumador. Una mezcla de óxidos se emplea como vidrio de pulimentar, y se añaden asimismo diferentes óxidos al vidrio para producir ciertas propiedades deseables. Algunas mezclas de europio e itrio se emplean como fósforo sensible al rojo en los televisores de color, etcétera.
Los actínidos
Como una recompensa a los químicos y físicos por descifrar el misterio de las tierras raras, los nuevos conocimientos proporcionaron la clave de la Química de los elementos situados al final de la tabla periódica, incluyendo los creados por el hombre.
Esta serie de elementos pesados empiezan con el actinio, número 89. En la tabla está situado debajo del lantano. El actinio tiene 2 electrones en la capa Q, del mismo modo que el lantano tiene otros 2 en la capa P. El electrón 89 y último del actinio pasa a ocupar la capa P, del mismo modo que el 57 y último del lantano ocupa la capa O. Ahora se plantea este interrogante: Los elementos situados detrás del actinio, ¿siguen añadiendo electrones a la capa P y convirtiéndose así en elementos usuales de transición? ¿O, por el contrario, se comportan como los elementos situados detrás del lantano, cuyos electrones descienden para completar la subcapa omitida situada debajo? Si ocurre esto, el actinio puede ser el comienzo de una nueva serie de «metales de tierras raras».
Los elementos naturales de esta serie son el actinio, el torio, el protactinio y el uranio. No fueron ampliamente estudiados hasta 1940. Lo poco que se sabía sobre su química sugería que se trataba de elementos usuales de transición. Pero cuando se añadieron a la lista los elementos neptunio y plutonio —elaborados por el hombre— y se estudiaron detenidamente, mostraron un gran parecido químico con el uranio. Glenn Seaborg fue urgido a proponer que los elementos pesados estaban, de hecho, siguiendo la pauta lantánida y llenando y llenando el cuarto subhueco sin llenar del hueco O. Con el laurencio se ha ocupado este subhueco, y los quince actínidos existen, en perfecta analogía con los quince lantánidos. Una confirmación importante es que la cromatografía de intercambios de iones separa los actínidos en la misma forma que separa los lantánidos.
Los elementos 104 (ruterfordio) y 105 (hahnio) son transactínidos y, los químicos están del todo seguros al respecto, aparecen por debajo del hafnio y tantalio, los dos elementos que siguen a los lantánidos.
Los gases
Desde los comienzos de la Química se reconoció que podían existir muchas sustancias en forma de gas, líquido o sólido, según la temperatura. El agua es el ejemplo más común: a muy baja temperatura se transforma en hielo sólido, y si se calienta mucho, en vapor gaseoso. Van Helmont —el primero en emplear la palabra «gas»— recalcó la diferencia que existe entre las sustancias que son gases a temperaturas usuales, como el anhídrido carbónico, y aquellas que, al igual que el vapor, son gases sólo a elevadas temperaturas. Llamó «vapores» a estos últimos, por lo cual seguimos hablando «de vapor de agua», no de «gas de agua».
El estudio de los gases o vapores siguió fascinando a los químicos, en parte porque les permitía dedicarse a estudios cuantitativos. Las leyes que determinan su conducta son más simples y fáciles de establecer que las que gobiernan el comportamiento de los líquidos y los sólidos.
Licuefacción
En 1787, el físico francés Jacques-Alexandre-César Charles descubrió que, cuando se enfriaba un gas, cada grado de enfriamiento determinaba una contracción de su volumen aproximadamente igual a 1/273 del volumen que el mismo gas tenía a 0 °C, y, a la inversa, cada grado de calentamiento provocaba una expansión del mismo valor. La expansión debida al calor no planteaba dificultades lógicas; pero si continuaba la disminución de volumen de acuerdo con la ley de Charles (tal como se la conoce hoy), al llegar a los –273 °C, el gas desaparecería. Esta paradoja no pareció preocupar demasiado a los químicos, pues se daban cuenta de que la ley de Charles no podía permanecer inmutable hasta llegar a temperaturas tan bajas, y, por otra parte, no tenían medio alguno de conseguir temperaturas lo suficientemente bajas como para ver lo que sucedía.
El desarrollo de la teoría atómica —que describía los gases como grupos de moléculas— presentó la situación en unos términos completamente nuevos. Entonces empezó a considerarse que el volumen dependía de la velocidad de las moléculas. Cuanto más elevada fuese la temperatura, a tanta mayor velocidad se moverían, más «espacio necesitarían para moverse» y mayor sería el volumen. Por el contrario, cuanto más baja fuese la temperatura, más lentamente se moverían, menos espacio necesitarían y menor sería el volumen. En la década de 1860, el físico británico William Thomson —que alcanzó la dignidad de par, como Lord Kelvin— sugirió que el contenido medio de energía de las moléculas era lo que disminuía en un índice del 1/273 por cada grado de enfriamiento. Si bien no podía esperarse que el volumen desapareciera por completo, la energía sí podía hacerlo. Según Thomson, a –273 °C, la energía de las moléculas descendería hasta cero, y éstas permanecerían inmóviles. Por tanto, –273 °C debe de ser la temperatura más baja posible. Así, pues, esta temperatura (establecida actualmente en –273,16 °C, según mediciones más modernas) sería el «cero absoluto», o, como se dice a menudo, el «cero Kelvin». En esta escala absoluta, el punto de fusión del hielo es de 273 K. En la figura 6.5 se representan las escalas Fahrenheit, centígrada y Kelvin.

Fig. 6.5. Comparación de las escalas termométricas Fahrenheit, Centígrada y Kelvin.
Este punto de vista hace aún más seguro que los gases se licuefactarían a medida que se aproximase el cero absoluto. Con incluso menos energía disponible, las moléculas de gas requerirían tan poco sitio que se colapsarían unas en otras y entrarían en contacto. En otras palabras, se convertirían en líquidos, puesto que las propiedades de los líquidos se explican al suponer que consisten en moléculas en líquido, pero que las moléculas contienen aún energía suficiente como para deslizarse y quedar libres por encima, por debajo y para pasarse unas a otras. Por esta razón, los líquidos pueden verterse y cambiar fácilmente de forma para adaptarse a un recipiente en particular.
A medida que la energía continúa decreciendo con el descenso de la temperatura, las moléculas llegarán a poseer tan poco espacio para abrirse paso las unas a las otras, e incluso para ocupar alguna posición fijada en la que vibrar, que no pueden moverse. En otras palabras, el líquido se ha helado y convertido en sólido. Así le pareció claro a Kelvin que, a medida que uno se aproximaba al cero absoluto, todos los gases no sólo se licuefactarían sino que se congelarían.
Naturalmente, entre los químicos existía un deseo de demostrar lo exacto de la sugerencia de Kelvin, haciendo descender la temperatura hasta el punto donde todos los gases se licuefactarían en primer lugar, y luego se congelarían, de la forma en que se consigue el cero absoluto. (Existe aquí algo respecto de un horizonte lejano que llama para su conquista.)
Los científicos habían estado explorando los extremos del frío mucho antes de que Kelvin hubiese definido el último objetivo. Michael Faraday descubrió que, incluso a temperaturas ordinarias, algunos gases se licuefactan bajo presión. Empleó un fuerte tubo de vidrio inclinado en forma de bumerán. En el extremo cerrado, colocó una sustancia que podría contener el gas tras el que iba. Luego, cerró el extremo abierto. El extremo con la materia sólida lo situó en agua caliente, con lo que se liberó el gas en una cantidad cada vez más creciente, y, puesto que el gas se hallaba confinado dentro del tubo, desarrolló una presión cada vez mayor. El otro extremo del tubo lo mantuvo Faraday en un cubilete lleno de hielo picado. En ese extremo, el gas se hallaría sometido tanto a una presión elevada como a baja temperatura, y se licuaría. En 1823, Faraday licuefacto de esta forma el gas cloro. El punto normal de licuefacción del cloro es de –34,5 °C (238,7 K).
En 1835, un químico francés, C. S. A. Thilorier, empleó el método de Faraday para formar dióxido de carbono líquido bajo presión, empleando cilindros metálicos, que podrían soportar mayores presiones que los tubos de vidrio. Preparó dióxido de carbono líquido en considerable cantidad y luego lo dejó escapar del tubo a través de una estrecha boquilla.
Naturalmente, en esas condiciones, el dióxido de carbono líquido, expuesto a las temperaturas normales se evaporaría con rapidez. Cuando un líquido se evapora, sus moléculas se separan de aquellas de las que está rodeado, a fin de convertirse en entidades singulares que se muevan con libertad. Las moléculas de un líquido tienen una fuerza de atracción entre sí, y el conseguir liberarse de la atracción es algo que requiere energía. Si la evaporación es rápida, no hay tiempo para que suficiente energía (en forma de calor) entre en el sistema, y la única restante fuente de energía para alimentar la evaporación es el líquido en sí. Por tanto, cuando el líquido se evapora rápidamente, la temperatura del líquido residual disminuye.
(Este fenómeno es algo experimentado por nosotros, puesto que el cuerpo humano siempre transpira ligeramente, y la evaporación de la fina capa de agua de nuestra piel retira calor de esa piel y nos mantiene frescos. Cuanto más calor hace, como es natural, más sudamos, y si el aire es húmedo y esa evaporación no puede tener lugar, la transpiración se recoge en nuestro cuerpo y nos llegamos a sentir incluso incómodos. El ejercicio, al multiplicar las reacciones productoras de calor dentro de nuestro cuerpo, también incrementa la transpiración, y asimismo nos encontramos incómodos en condiciones de humedad.)
Cuando Thilorier (para volver a él) permitió al dióxido de carbono evaporarse, la temperatura del líquido descendió a medida que tenía lugar la evaporación, hasta que el dióxido de carbono se congeló. Por primera vez se había conseguido formar dióxido de carbono sólido.
El dióxido de carbono líquido es estable sólo bajo presión. El dióxido de carbono sólido expuesto a condiciones ordinarias se sublima, es decir, se evapora directamente a gas sin fundirse. El punto de sublimación del dióxido de carbono sólido es de –78,5 °C (194,7 K).
El dióxido de carbono sólido tiene la apariencia de hielo empañado (aunque está mucho más frío), y dado que no forma un líquido se le ha llamado hielo seco. Cada año se producen unas 400.000 toneladas del mismo, y la mayor parte se emplea para preservar los alimentos a través de la refrigeración.
El enfriamiento por evaporación revolucionó la vida humana. Con anterioridad al siglo XIX, el hielo, cuando estaba disponible, podía emplearse para conservar los alimentos. El hielo debía recogerse en invierno y guardarse, bajo aislamiento, durante el verano; o bien había que bajarlo desde las montañas. En el mejor de los casos era un proceso tedioso y difícil, y la mayoría de la gente debía improvisarlo en verano (o con el calor de todo el año, pongamos por caso).
Ya en 1755, el químico escocés William Cullen había producido hielo al formar un vacío sobre ciertas cantidades de agua, ayudando a la rápida evaporación, que enfriaba el agua hasta el punto de congelación. Naturalmente, esto no podía competir con el hielo natural. Ni tampoco el proceso podía usarse indirectamente de una forma simple para enfriar los alimentos, puesto que se formaría hielo y obstruiría los conductos.
En la actualidad, un gas apropiado se licuefacta con un compresor y se le hace alcanzar la temperatura deseada. A continuación se le obliga a circular por un serpentín en torno a una cámara en donde se encuentran los alimentos. A medida que se evapora, retira el calor de la cámara. El gas que sale es de nuevo licuefactado por un compresor, se le enfría y se le hace circular. El proceso es continuo, y el calor es bombeado fuera de la cámara cerrada hasta la atmósfera exterior. El resultado es un frigorífico, que ha remplazado a las viejas neveras de hielo.
En 1834, un inventor norteamericano, Jacob Perkins, patentó (en Gran Bretaña) el empleo del éter como refrigerante. Otros gases, como el amoníaco y el dióxido de azufre empezaron también a emplearse. Todos estos refrigerantes tenían la desventaja de ser venenosos o inflamables. Sin embargo, en 1930, el químico estadounidense Thomas Midgley descubrió el diclorodifluormetano (CF2Cl2), mucho más conocido bajo su nombre registrado de «Freón». No es tóxico (como demostró Midgley llenándose de él los pulmones en público), es ininflamable y se adecúa a la perfección a estas funciones. Con el freón, la refrigeración doméstica se convirtió en algo extendido y popular.
(Aunque el freón y otros fluorocarbonos han demostrado en todo momento ser inofensivos para los seres humanos, las dudas comenzaron a presentarse, en la década de 1970, respecto a su efecto sobre la ozonosfera, tal y como se ha descrito en el capítulo anterior.)
La refrigeración también se aplica, moderadamente, en unos volúmenes mayores en el acondicionamiento del aire, así llamado porque el aire se acondiciona, es decir, se filtra y se deshumidifica. La primera unidad práctica de acondicionamiento del aire se diseñó en 1902 por el inventor norteamericano Willis Haviland Carrier y, después de la Segunda Guerra Mundial, el aire acondicionado se ha convertido en algo ampliamente difundido en las ciudades norteamericanas y de otros países.
Volvamos de nuevo a Thilorier. Añadió dióxido de carbono sólido en un líquido llamado dietiléter (más conocido hoy como anestésico, véase capítulo 11). El dietiléter tiene un punto bajo de ebullición y se evapora con rapidez. Entre él y la baja temperatura del dióxido de carbono sólido, que se sublima, se consigue una temperatura de –110 °C (163,2 K).
En 1845, Faraday volvió a la tarea de licuefactar gases bajo el efecto combinado de la baja temperatura y de la alta presión, empleando dióxido de carbono sólido y el dietiléter como mezcla enfriante. A pesar de esta mezcla y del empleo de unas presiones más elevadas que antes, había seis gases que no se licuefactaban. Se trataba del hidrógeno, el oxígeno, el nitrógeno, el monóxido de carbono, el óxido nítrico y el metano, por lo que les llamó gases permanentes. A esta lista podemos añadir cinco gases más que Faraday no conocía en su época. Uno de ellos era el flúor, y los otros cuatro son los gases nobles: helio, neón, argón y criptón.
Sin embargo, en 1869 el físico irlandés Thomas Andrews dedujo a partir de sus experimentos que todo gas poseía una temperatura crítica por encima de la cual no se podía licuefactar, ni siquiera bajo presión. Esta presunción fue más tarde vertida en una firme base teórica por el físico neerlandés Johannes van der Waals quien, como resultado de todo esto, consiguió el premio Nobel de Física en 1910.
Por lo tanto, para licuefactar cualquier gas se ha de estar seguro de que se trabaja a unas temperaturas por debajo del valor crítico, o en otro caso todo constituirá algo baldío. Se hicieron grandes esfuerzos para alcanzar unas temperaturas aún más bajas para conquistar a estos tozudos gases. Un método en cascada —bajar la temperatura paso a paso— demostró ser el truco mejor. En primer lugar, el dióxido de azufre licuefactado, enfriado a través de la evaporación, se empleó para licuefactar el dióxido de carbono; a continuación, el dióxido de carbono líquido se empleó para licuar un gas más resistente, etcétera. En 1977, el físico suizo Raoul Pictet consiguió finalmente licuefactar oxígeno, a una temperatura de –140 °C (133 K) y a una presión de 500 atmósferas (21.000 kg/cm2). El físico francés Louis Paul Caitellet, aproximadamente en la misma época, licuefacto no sólo el oxígeno sino también el nitrógeno y el monóxido de carbono. Naturalmente, esos líquidos hicieron posible el seguir adelante y el conseguir unas temperaturas aún más bajas. El punto de licuefacción del oxígeno a la presión ordinaria del aire se demostró a su debido tiempo que era la de –183 °C (90 K), la del monóxido de carbono, –190 °C (83 K), y la del nitrógeno, –195 °C (78 K).
En 1895, el ingeniero químico inglés William Hampson y el físico alemán Karl von Linde, independientemente, idearon una forma de licuefactar el aire a gran escala. El aire era primero comprimido y enfriado a temperaturas ordinarias. Luego se le dejaba expansionar y, en el proceso, quedaba por completo helado. Este aire helado se empleaba para bañar un contenedor de aire comprimido hasta que quedaba del todo frío. El aire comprimido se dejaba expansionar a continuación, con lo que se volvía aún más frío. Se repitió el proceso, consiguiendo cada vez un aire más y más frío, hasta que se licuefacto.
El aire líquido, en gran cantidad y barato, se separó con facilidad en oxígeno y nitrógeno líquidos. El oxígeno podía emplearse en lámparas de soldar y para fines médicos; el nitrógeno, también resultaba útil en estado inerte.
Así, las lámparas de incandescencia llenas de nitrógeno permitieron que los filamentos permaneciesen a unas temperaturas al rojo blanco durante mayores períodos de tiempo, antes de que la lenta evaporación del metal las destruyese, que de haber estado los mismos filamentos en bombillas a las que se hubiese hecho el vacío. El aire líquido podía emplearse asimismo como fuente para unos componentes menores, tales como el argón y otros gases nobles.
El hidrógeno resistió todos los esfuerzos para licuefactarlo hasta 1900. El químico escocés James Dewar llevó a cabo esta proeza al recurrir a una nueva estratagema. Lord Kelvin (William Thomson) y el físico inglés James Prescott Joule habían mostrado que, incluso en estado gaseoso, un gas podía enfriarse, simplemente, permitiéndole expansionarse e impidiendo que el calor se filtre en el gas desde el exterior, siempre y cuando la temperatura sea lo suficientemente baja como para iniciar el proceso. Entonces, Dewar enfrió el hidrógeno comprimido hasta una temperatura de –200 °C en una vasija rodeada de nitrógeno líquido, dejando que este superfrígido hidrógeno se expansionase y se enfriara aún más, repitiendo el ciclo una y otra vez, haciendo pasar el hidrógeno cada vez más frío por unos conductos. El hidrógeno comprimido, sometido a este efecto Joule-Thomson, se hizo finalmente líquido a una temperatura de unos –240 °C (33 K). A temperaturas aún inferiores, Dewar consiguió obtener hidrógeno sólido.
Para conservar estos líquidos superenfriados, ideó unos frascos especiales revestidos de plata. Se les dotó de una doble pared en medio de la cual se había hecho el vacío. El calor no se pierde (o se gana) a través del vacío más que con ayuda de un proceso comparativamente lento de radiación, y el revestimiento de plata reflejaba la radiación entrante (o, pongamos por caso, saliente). Esos frascos Dewar son el antepasado directo de los termos domésticos.
Combustible de cohetes
Con la llegada de los cohetes, los gases licuefactados consiguieron niveles aún mayores de popularidad. Los cohetes requerían una reacción química en extremo rápida, que contuviese grandes cantidades de energía. El tipo más conveniente de combustible era uno líquido, como el alcohol o el queroseno, y oxígeno líquido. El oxígeno, o algún agente oxidante alternativo, debe llevarse en el cohete en todo caso, porque el cohete queda desprovisto de cualquier suplemento natural de oxígeno cuando abandona la atmósfera. Y el oxígeno debe encontrarse en forma líquida, puesto que los líquidos son más densos que los gases, y puede albergarse en los depósitos de combustible más oxígeno en forma líquida que en forma gaseosa. Por consiguiente, el oxígeno líquido ha sido muy solicitado para todo lo relacionado con los cohetes.
La efectividad de una mezcla de combustible y oxidante se mide por el llamado «impulso específico» el cual representa el número de kilos de empuje producidos por la combustión de 1 kg de la mezcla de combustible-oxidante por segundo. Para una mezcla de queroseno y oxígeno, el impulso específico es igual a 121. Puesto que la carga máxima que un cohete puede transportar depende del impulso específico, se buscaron combinaciones más eficaces. Desde este punto de vista, el mejor combustible químico es el hidrógeno líquido. Combinado con oxígeno líquido, puede dar un impulso específico igual a 175 aproximadamente. Si el ozono o el flúor líquidos pudiesen usarse igual que el oxígeno, el impulso específico podría elevarse hasta 185.
Ciertos metales ligeros, como el litio, el boro, el magnesio, el aluminio y, particularmente, el berilio, liberan más energía o se combinan con más oxígeno de lo que hace incluso el hidrógeno. Sin embargo, algunos de ellos son raros, y todos implican dificultades técnicas en su quemado, dificultades provenientes de la carencia de humos, de los depósitos de óxido, etcétera.
También existen combustibles sólidos que hacen las veces de sus propios oxidantes (como la pólvora, que fue el primer propelente de los cohetes, pero mucho más eficiente). Tales combustibles se denominan monopropelentes, puesto que no necesitan recurrir al suplemento o al oxidante y son por sí mismos el propelente requerido. Los combustibles que también precisan de oxidantes son los bipropelentes. Los monopropelentes se guardan y utilizan con facilidad, y arden de una forma rápida pero controlada. La dificultad principal radica, probablemente, en desarrollar un monopropelente con un impulso específico que se aproxime al de los bipropelentes.
Otra posibilidad la constituye el hidrógeno atómico, como el que empleó Langmuir en su soplete. Se ha calculado que el motor de un cohete que funcionase mediante la recombinación de átomos de hidrógeno para formar moléculas, podría desarrollar un impulso específico de más de 650. El problema principal radica en cómo almacenar el hidrógeno atómico. Hasta ahora, lo más viable parece ser un rápido y drástico enfriamiento de los átomos libres, inmediatamente después de formarse éstos. Las investigaciones realizadas en el «National Bureau of Standards» parecen demostrar que los átomos de hidrógeno libre quedan mejor preservados si se almacenan en un material sólido a temperaturas extremadamente bajas —por ejemplo, oxígeno congelado o argón—. Si se pudiese conseguir que apretando un botón —por así decirlo— los gases congelados empezasen a calentarse y a evaporarse, los átomos de hidrógeno se liberarían y podrían recombinarse. Si un sólido de este tipo pudiese conservar un 10 % de su peso en átomos libres de hidrógeno, el resultado sería un combustible mejor que cualquiera de los que poseemos actualmente. Pero, desde luego, la temperatura tendría que ser muy baja, muy inferior a la del hidrógeno líquido. Estos sólidos deberían ser mantenidos a temperaturas de –272 °C, es decir, a un solo grado por encima del cero absoluto.
Otra solución radica en la posibilidad de impulsar los iones en sentido retrógrado (en vez de los gases de salida del combustible quemado). Cada uno de los iones, de masa pequeñísima, produciría impulsos pequeños, pero continuados, durante largos períodos de tiempo. Así, una nave colocada en órbita por la fuerza potente —aunque de breve duración— del combustible químico, podría, en el espacio —medio virtualmente libre de fricción—, ir acelerando lentamente, bajo el impulso continuo de los iones, hasta alcanzar casi la velocidad de la luz. El material más adecuado para tal impulso iónico es el cesio, la sustancia que más fácilmente puede ser forzada a perder electrones y a formar el ion de cesio. Luego puede crearse un campo eléctrico para acelerar el ion de cesio y dispararlo por el orificio de salida del cohete.
Superconductores y superfluidos
Pero volvamos al mundo de las bajas temperaturas. Ni siquiera la licuefacción y la solidificación del hidrógeno constituyen la victoria final. En el momento en que se logró dominar el hidrógeno, se habían descubierto ya los gases inertes, el más ligero de los cuales, el helio, se convirtió en un bastión inexpugnable contra la licuefacción a las más bajas temperaturas obtenibles. Finalmente, en 1908, el físico holandés Heike Kammerlingh Onnes consiguió dominarlo. Dio un nuevo impulso al sistema Dewar. Empleando hidrógeno líquido, enfrió bajo presión el gas de helio hasta –255 °C aproximadamente y luego dejó que el gas se expandiese para enfriarse aún más. Este método le permitió licuar el gas. Luego, dejando que se evaporase el helio líquido, consiguió la temperatura a la que podía ser licuado el helio bajo una presión atmosférica normal, e incluso a temperaturas de hasta –272,3 °C. Por su trabajo sobre las bajas temperaturas, Onnes recibió el premio Nobel de Física en 1913. (Hoy es algo muy simple la licuefacción del helio. En 1947, el químico americano Samuel Cornette Collins inventó el «criostato», con el cual, por medio de compresiones y expansiones alternativas, puede producir hasta 34 litros de helio líquido por hora.) Sin embargo, Onnes hizo mucho más que obtener nuevos descensos en la temperatura. Fue el primero en demostrar que a estos niveles existían propiedades únicas de la materia.
Una de estas propiedades es el extraño fenómeno denominado «superconductividad». En 1911, Onnes estudió la resistencia eléctrica del mercurio a bajas temperaturas. Esperaba que la resistencia a una corriente eléctrica disminuiría constantemente a medida que la desaparición del calor redujese las vibraciones normales de los átomos en el metal. Pero a –268,88 °C desapareció súbitamente la resistencia eléctrica del mercurio. Una corriente eléctrica podía cruzarlo sin pérdida alguna de potencia. Pronto se descubrió que otros metales podían también transformarse en superconductores. Por ejemplo, el plomo lo hacía a –265,78 °C. Una corriente eléctrica de varios centenares de amperios —aplicada a un anillo de plomo mantenido en dicha temperatura por medio del helio líquido— siguió circulando a través de este anillo durante dos años y medio, sin pérdida apreciable de intensidad.
A medida que descendían las temperaturas, se iban añadiendo nuevos metales a la lista de los materiales superconductores. El estaño se transformaba en superconductor a los –269,27 °C; el aluminio, a los –271,80 °C; el uranio, a los –272,2 °C; el titanio, a los –272,47 °C; el hafnio, a los –272,65 °C. Pero el hierro, níquel, cobre, oro, sodio y potasio deben de tener un punto de transición mucho más bajo aún —si es que realmente pueden ser transformados en superconductores—, porque no se han podido reducir a este estado ni siquiera a las temperaturas más bajas alcanzadas. El punto más alto de transición encontrado para un metal es el del tecnecio, que se transforma en superconductor por debajo de los –261,8 °C.
Un líquido de bajo punto de ebullición retendrá fácilmente las sustancias inmersas en él a su temperatura de ebullición. Para conseguir temperaturas inferiores se necesita un líquido cuyo punto de ebullición sea aún menor. El hidrógeno líquido hierve a -252,6 °C, y sería muy útil encontrar una sustancia superconductora cuya temperatura de transición fuera, por lo menos, equivalente. Sólo tales condiciones permiten estudiar la superconductividad en sistemas refrigerados por el hidrógeno líquido. A falta de ellas, será preciso utilizar, como única alternativa, un líquido cuyo punto de ebullición sea bajo, por ejemplo, el helio líquido, elemento mucho más raro, más costoso y de difícil manipulación. Algunas aleaciones, en especial las que contienen niobio, poseen unas temperaturas de transición más elevadas que las de cualquier metal puro. En 1968 se encontró, por fin, una aleación de niobio, aluminio y germanio, que conservaba la superconductividad a –252 °C. Esto hizo posible la superconductividad a temperaturas del hidrógeno líquido, aunque con muy escaso margen. E inmediatamente se presentó, casi por sí sola, una aplicación útil de la superconductividad en relación con el magnetismo. Una corriente eléctrica que circula por un alambre arrollado en una barra de hierro, crea un potente campo magnético; cuanto mayor sea la corriente, tanto más fuerte será el campo magnético. Por desgracia, también cuanto mayor sea la corriente, tanto mayor será el calor generado en circunstancias ordinarias, lo cual limita considerablemente las posibilidades de tal aplicación. Ahora bien, la electricidad fluye sin producir calor en los alambres superconductores, y, al parecer, en dichos alambres se puede comprimir la corriente eléctrica para producir un «electroimán» de potencia sin precedentes con sólo una fracción de la fuerza que se consume en general. Sin embargo, hay un inconveniente.
En relación con el magnetismo, se ha de tener en cuenta otra característica, además de la superconductividad. En el momento en que una sustancia se transforma en superconductora, se hace también perfectamente «diamagnética», es decir, excluye las líneas de fuerza de un campo magnético. Esto fue descubierto por W. Meissner en 1933, por lo cual se llama desde entonces «efecto Meissner». No obstante, si se hace el campo magnético lo suficientemente fuerte, puede destruirse la superconductividad de la sustancia, incluso a temperaturas muy por debajo de su punto de transición. Es como si, una vez concentradas en los alrededores las suficientes líneas de fuerza, algunas de ellas lograran penetrar en la sustancia y desapareciese la superconductividad.
Se han realizado varias pruebas con objeto de encontrar sustancias superconductoras que toleren potentes campos magnéticos. Por ejemplo, hay una aleación de estaño y niobio con una elevada temperatura de transición: –255 °C. Puede soportar un campo magnético de unos 250.000 gauss, lo cual, sin duda, es una intensidad elevada. Aunque este descubrimiento se hizo en 1954, hasta 1960 no se perfeccionó el procedimiento técnico para fabricar alambres con esta aleación, por lo general, quebradiza. Todavía más eficaz es la combinación de vanadio y galio, y se han fabricado electroimanes superconductores con intensidades de hasta 500.000 gauss.
En el helio se descubrió también otro sorprendente fenómeno a bajas temperaturas: la «superfluidez».
El helio es la única sustancia conocida que no puede ser llevada a un estado sólido, ni siquiera a la temperatura del cero absoluto. Hay un pequeño contenido de energía irreductible, incluso al cero absoluto, que, posiblemente, no puede ser eliminada (y que, de hecho, su contenido en energía es «cero»); sin embargo, basta para mantener libres entre sí los extremadamente «no adhesivos» átomos de helio y, por tanto, líquidos. En 1905, el físico alemán Hermann Walther Nernst demostró que no es la energía de las sustancias la que se convierte en cero en el cero absoluto, sino una propiedad estrechamente vinculada a la misma: la «entropía». Esto valió a Nernst el premio Nobel de Química en 1920. Sea como fuere, esto no significa que no exista helio sólido en ninguna circunstancia. Puede obtenerse a temperaturas inferiores a 0,26 K y a una presión de 25 atmósferas aproximadamente.
En 1935, Willem Hendrik Keeson y su hermana, que trabajaban en el «Laboratorio Onnes», de Leiden, descubrieron que, a la temperatura de –270,8 °C el helio líquido conducía el calor casi perfectamente. Y lo conduce con tanta rapidez, que cada una de las partes de helio está siempre a la misma temperatura. No hierve —como lo hace cualquier otro líquido en virtud de la existencia de áreas puntiformes calientes, que forman burbujas de vapor— porque en el helio líquido no existen tales áreas (si es que puede hablarse de las mismas en un líquido cuya temperatura es de menos de –271 °C). Cuando se evapora, la parte superior del líquido simplemente desaparece, como si se descamara en finas láminas, por así decirlo.
El físico ruso Peter Leonidovich Kapitza siguió investigando esta propiedad y descubrió que si el helio era tan buen conductor del calor se debía al hecho de que fluía con notable rapidez y transportaba casi instantáneamente el calor de una parte a otra de sí mismo (por lo menos, doscientas veces más rápido que el cobre, el segundo mejor conductor del calor). Fluiría incluso más fácilmente que un gas, tendría una viscosidad de sólo 1/1.000 de la del hidrógeno gaseoso y se difundiría a través de unos poros tan finos, que podrían impedir el paso de un gas. Más aún, este líquido superfluido formaría una película sobre el cristal y fluiría a lo largo de éste tan rápidamente como si pasase a través de un orificio. Colocando un recipiente abierto, que contuviera este líquido, en otro recipiente mayor, pero menos lleno, el fluido rebasaría el borde del cristal y se desplazaría al recipiente exterior, hasta que se igualaran los niveles de ambos recipientes.
El helio es la única sustancia que muestra este fenómeno de superfluidez. De hecho, el superfluido se comporta de forma tan distinta al helio cuando está por encima de los -270,8 °C que se le ha dado un nombre especial: helio II, para distinguirlo del helio líquido cuando se halla por encima de dicha temperatura, y que se denomina helio I.
Sólo el helio permite investigar las temperaturas cercanas al cero absoluto, por lo cual se ha convertido en un elemento muy importante, tanto en la ciencias puras como en las aplicadas. La cantidad de helio que contiene la atmósfera es despreciable, y las fuentes más importantes son los pozos de gas natural, de los cuales escapa a veces el helio, formado a partir de la desintegración del uranio y el torio en la corteza terrestre. El gas del pozo más rico que se conoce (en Nuevo México) contiene un 7,5 % de helio.
Criogenia
Sorprendidos por los extraños fenómenos descubiertos en las proximidades del cero absoluto, los físicos, naturalmente, han realizado todos los esfuerzos imaginables por llegar lo más cerca posible del mismo y ampliar sus conocimientos acerca de lo que hoy se conoce con el nombre de «criogenia». En condiciones especiales, la evaporación del helio líquido puede dar temperaturas de hasta –272,5 °C. (Tales temperaturas se miden con ayuda de métodos especiales, en los cuales interviene la electricidad, así, por la magnitud de la corriente generada en un par termoeléctrico; la resistencia de un cable hecho de algún metal no superconductor; los cambios en las propiedades magnéticas, e incluso la velocidad del sonido del helio. La medición de temperaturas extremadamente bajas es casi tan difícil como obtenerlas.) Se han conseguido temperaturas sustancialmente inferiores a los –272,5 °C gracias a una técnica que empleó por primera vez, en 1925, el físico holandés Peter Joseph Wilhelm Debye. Una sustancia «paramagnética» —es decir, que concentra las líneas de fuerza magnética— se pone casi en contacto con el helio líquido, separado de éste por gas de helio, y la temperatura de todo el sistema se reduce hasta –272 °C. Luego se coloca el sistema en un campo magnético. Las moléculas de la sustancia paramagnética se disponen paralelamente a las líneas del campo de fuerza, y al hacerlo desprenden calor, calor que se extrae mediante una ligera evaporación del helio ambiente. Entonces se elimina el campo magnético. Las moléculas paramagnéticas adquieren inmediatamente una orientación arbitraria. Al pasar de una orientación ordenada a otra arbitraria, las moléculas han de absorber calor, y lo único que puede hacer es absorberlo del helio líquido. Por tanto, desciende la temperatura de éste.
Esto puede repetirse una y otra vez, con lo cual desciende en cada ocasión la temperatura del helio líquido. La técnica fue perfeccionada por el químico americano William Francis Giauque, quien recibió el premio Nobel de Química en 1949 por estos trabajos. Así, en 1957 se alcanzó la temperatura de –272,99998 °C.
En 1962, el físico germano-británico Heinz London y sus colaboradores creyeron posible emplear un nuevo artificio para obtener temperaturas más bajas aún. El helio se presenta en dos variantes: helio 3 y helio 4. Por lo general, ambas se mezclan perfectamente, pero cuando las temperaturas son inferiores a los –272,2 °C, más o menos, los dos elementos se disocian, y el helio 3 sobrenada. Ahora bien, una porción de helio 3 permanece en la capa inferior con el helio 4, y entonces se puede conseguir que el helio 3 suba y baje repetidamente a través de la divisoria, haciendo descender cada vez más la temperatura, tal como ocurre con los cambios de liquidación y vaporización en el caso de refrigerantes corrientes tipo freón. En 1965 se construyeron en la Unión Soviética los primeros aparatos refrigeradores en los que se aplicó este principio.
En 1950, el físico ruso Isaak Yakovievich Pomeranchuk propuso un método de refrigeración intensa, en el que se aplicaban otras propiedades del helio 3. Por su parte, el físico británico de origen húngaro, Nicholas Kurti, sugirió, ya en 1934, el uso de propiedades magnéticas similares a las que aprovechara Giauque, si bien circunscribiendo la operación al núcleo atómico —la estructura más recóndita del átomo—, es decir, prescindiendo de las moléculas y los átomos completos.
Como resultado del empleo de esas nuevas técnicas, han llegado a conseguir temperaturas tan bajas como de 0,000001 K. Y, dado que los físicos se encuentran a una millonésima de grado del cero absoluto, ¿no podrían desembarazarse de la pequeña entropía que queda y, finalmente, llegar a la marca del 0 K absoluto?
¡No! El cero absoluto es inalcanzable, como demostró Nernst a través de su premio Nobel ganado al tratar de este tema (en ocasiones se denomina a esto tercera ley de la termodinámica). En cualquier descenso de temperatura, sólo parte de la entropía puede eliminarse. En general, eliminar la mitad de la entropía de un sistema es igualmente difícil, sin tener en cuenta cuál sea su total. Así resulta tan difícil avanzar desde los 300 K (más o menos la temperatura ambiente) hasta los 150 K (algo más frío que cualquier temperatura reinante en la Antártida), que desde 20 K a 10 K. Resulta igual de difícil avanzar desde los 10 K hasta los 5 K, y desde los 5 K hasta los 2,5 K, etc. Al haber alcanzado una millonésima de grado por encima del cero absoluto, la tarea de avanzar más allá, hasta la mitad de una millonésima de grado resulta tan difícil como el bajar de 300 K a 150 K, y si se consiguiese, sería una tarea igualmente difícil el avanzar desde media millonésima de grado hasta una cuarta parte de millonésima de grado, y así indefinidamente. El cero absoluto se encuentra a una distancia infinita, sin tener en cuenta lo cerca que parezca que nos aproximamos.
Digamos a este respecto que los estadios finales de la búsqueda del cero absoluto se han conseguido a partir de un estudio atento del helio 3. El helio 3 es una sustancia en extremo rara. El helio en sí no es muy común en la Tierra, y cuando se aisló sólo 13 átomos de cada 10.000.000 son de helio 3, y el resto es helio 4.
El helio 3 es en realidad un átomo más simple que el helio 4, y sólo posee las tres cuartas partes de la masa de la variedad más frecuente. El punto de licuefacción del helio 3 es de 3,2 K, un grado más por debajo que el helio 4. Y lo que es más, al principio se creyó que, dado que el helio 4 se hace superfluido a temperaturas por debajo de 2,2 K, el helio 3 (una molécula menos simétrica, aunque sea más simpie) no muestra señales de superfluidez en absoluto. Era sólo necesario seguir intentándolo. En 1972, se descubrió que el helio 3 cambia a una forma de helio II superfluido líquido a temperaturas por debajo de 0,0025 K.
Altas presiones
Uno de los nuevos horizontes científicos abiertos por los estudios de la licuefacción de gases fue el desarrollo del interés por la obtención de altas presiones. Parecía que sometiendo a grandes presiones diversos tipos de materia (no sólo los gases), podría obtenerse una información fundamental sobre la naturaleza de la materia y sobre el interior de la Tierra. Por ejemplo, a una profundidad de 11 km, la presión es de 1.000 atmósferas; a los 643 km, de 200.000 atmósferas; a los 3.218 km de 1.400.000 atmósferas, y en el centro de la Tierra, a más de 6.400 km de profundidad, alcanza los 3.000.000 de atmósferas. (Por supuesto que la Tierra es un planeta más bien pequeño. Se calcula que las presiones centrales en Saturno son de más de 50 millones de atmósferas, y en el coloso Júpiter, de 100 millones.)
La presión más alta que podía obtenerse en los laboratorios del siglo XIX era de 3.000 atmósferas, conseguidas por E. H. Amagat en la década de 1880. Pero en 1905, el físico americano Percy Williams Bridgman empezó a elaborar nuevos métodos, que pronto permitieron alcanzar presiones de 20.000 atmósferas e hicieron estallar las pequeñas cámaras de metal que empleaba para sus experimentos. Siguió probando con materiales más sólidos, hasta conseguir presión de hasta más de 1.000.000 de atmósferas. Por sus trabajos en este campo, recibió el premio Nobel de Física en 1946.
Estas presiones ultraelevadas permitieron a Bridgman forzar a los átomos y moléculas de una sustancia a adoptar agrupaciones más compactas, que a veces se mantenían una vez eliminada la presión. Por ejemplo, convirtió el fósforo amarillo corriente —mal conductor de la electricidad— en fósforo negro, una forma de fósforo conductora. Logró sorprendentes cambios, incluso en el agua. El hielo normal es menos denso que el agua líquida. Utilizando altas presiones, Bridgman produjo una serie de hielos («hielo-II», «hielo-III», etc.), que no sólo eran más densos que el líquido, sino que eran hielo sólo a temperaturas muy por encima del punto normal de congelación del agua. El hielo-VII es un sólido a temperaturas superiores a la del punto de ebullición del agua.
La palabra «diamante» es la más sugestiva en el mundo de las altas presiones. Como sabemos, el diamante es carbón cristalizado, igual que el grafito. (Cuando aparece un elemento en dos formas distintas, estas formas se llaman «alótropas». El diamante y el grafito constituyen los ejemplos más espectaculares de este fenómeno. Otros ejemplos los tenemos en el ozono y el oxígeno corriente.) La naturaleza química del diamante la demostraron por vez primera, en 1772, Lavoisier y algunos químicos franceses colegas suyos. Compraron un diamante y procedieron a calentarlo a temperaturas lo suficientemente elevadas como para quemarlo. Descubrieron que el gas resultante era anhídrido carbónico. Más tarde, el químico británico Smithson Tennant demostró que la cantidad de anhídrido carbónico medida sólo podía obtenerse si el diamante era carbono puro, y, en 1799, el químico francés Guyton de Morveau puso punto final a la investigación convirtiendo un diamante en un pedazo de grafito.
Desde luego, se trataba de un mal negocio; pero, ¿por qué no podía realizarse el experimento en sentido contrario? El diamante es un 55 % más denso que el grafito. ¿Por qué no someter el grafito a presión y forzar a los átomos componentes a adoptar la característica disposición más densa que poseen en el diamante?
Se realizaron muchos esfuerzos, y, al igual que había sucedido con los alquimistas, varios investigadores informaron haber alcanzado éxito. El más famoso fue el químico francés Ferdinand-Frédéric-Henri Moissan. En 1893 disolvió grafito en hierro colado fundido, e informó que había encontrado pequeños diamantes en la masa después de enfriada. La mayor parte de los objetos eran negros, impuros y pequeños; pero uno de ellos era incoloro y medía casi un milímetro de largo. Estos resultados se aceptaron en general, y durante largo tiempo se creyó que Moissan había obtenido diamantes sintéticos. Pese a ello, nunca se repitieron con éxito sus resultados.
Sin embargo, la búsqueda de diamantes sintéticos proporcionó algunos éxitos marginales. En 1891, el inventor americano Edward Goodrich Acheson descubrió, durante sus investigaciones en este campo, el carburo de silicio, al que dio el nombre comercial de «Carborundo». Este material era más duro que cualquier sustancia conocida hasta entonces, a excepción del diamante, y se ha empleado mucho como abrasivo, es decir, que se trata de una sustancia usada para pulir y abrillantar.
La eficacia de un abrasivo depende de su dureza. Un abrasivo puede pulir o moler sustancias menos duras que él, y, en este aspecto, el diamante, como la sustancia más dura, es la más eficaz. La dureza de las diversas sustancias suele medirse por la «escala de Mohs», introducida, en 1818, por el minerólogo alemán Friedrich Mohs. Dicha escala asigna a los minerales números desde el 1 —para el talco— hasta el 10 — para el diamante—. Un mineral de un número determinado puede rayar todos los minerales de números más bajos que él. En la escala de Mohs, el carborundo tiene el número 9. Sin embargo, las divisiones no son iguales. En una escala absoluta, la diferencia de dureza entre el número 10 (diamante) y el 9 (carborundo) es cuatro veces mayor que la existente entre el 9 (carborundo) y el 1 (talco).
La razón de todo esto no resulta muy difícil de comprender. En el grafito, los átomos de carbono están dispuestos en capas. En cada capa individual, los átomos de carbono se hallan dispuestos en hexágonos en mosaico, como las baldosas del suelo de un cuarto de baño. Cada átomo de carbono está unido a otros tres de igual forma y, dado que el carbono es un átomo pequeño, los vecinos están muy cerca y fuertemente unidos. La disposición en mosaico es muy difícil de separar, pero es muy tenue y se rompe con facilidad. Una disposición en mosaico se halla a una distancia comparativamente grande en relación al siguiente mosaico, por encima y por debajo, por lo que los lazos entre las capas son débiles, y es posible hacer deslizar una capa por encima de la siguiente. Por esta razón, el grafito no es sólo particularmente fuerte sino que, en realidad, puede usarse como lubricante.
Sin embargo, en el diamante los átomos de carbono están dispuestos con una simetría absolutamente tridimensional. Cada átomo de carbono se halla enlazado a otros cuatro y a iguales distancias, y cada uno de los cuatro constituyen los ápices de un tetraedro en el que los átomos de carbono en consideración constituyen el centro. Esta disposición es muy compacta, por lo que, sustancialmente, el diamante es más denso que el grafito. No puede separarse en ninguna dirección, excepto bajo una fuerza enorme. Otros átomos pueden adoptar la configuración diamantina, pero de ellos el átomo de carbono es el más pequeño y el que se mantiene más fuertemente unido. Así, el diamante es más duro que cualquier otra sustancia bajo las condiciones de la superficie terrestre.
En el carburo de silicio, la mitad de los átomos de carbono están sustituidos por átomos de silicio. Dado que los átomos de silicio son considerablemente más grandes que los átomos de carbono, no se unen a sus vecinos con tanta fuerza y sus enlaces son débiles. De este modo, el carburo de silicio no es tan duro como el diamante (aunque sea lo suficientemente fuerte para numerosos fines).
Bajo las condiciones de la superficie de la Tierra, la disposición de los átomos de carbono del grafito es más estable que la disposición del diamante. Por lo tanto, existe una tendencia a que el diamante se convierta espontáneamente en grafito. Sin embargo, no existe el menor peligro en que uno se despierte y se encuentre a su magnífico anillo de diamantes convertido en algo sin valor de la noche a la mañana. Los átomos de carbono, a pesar de su inestable disposición, se mantienen unidos con la fuerza suficiente como para que hagan falta muchísimos millones de años para que este cambio tenga lugar.
La diferencia en estabilidad hace aún más difícil el convertir al grafito en diamante. No fue hasta la década de los años 1930 cuando, finalmente, los químicos consiguieron los requisitos de presión necesarios para convertir el grafito en diamante. Se demostró que esta conversión necesitaba de una presión, por lo menos, de 10.000 atmósferas, y aun así era una cosa impracticablemente lenta. El elevar la temperatura aceleraría la conversión, pero también aumentaría los requisitos en la presión. A 1.500 °C, se necesita por lo menos una presión de 30.000 atmósferas. Todo esto probó que Moissan y sus contemporáneos, bajo las condiciones que empleaban, no hubieran podido producir diamantes del mismo modo que los alquimistas oro. (Existen algunas pruebas de que Moissan fue en realidad víctima de uno de sus ayudantes, el cual, para librarse de aquellos tediosos experimentos, decidió acabar con los mismos colocando un diamante auténtico en la mezcla de hierro fundido.)
Ayudado por el trabajo de pionero de Bridgman al conseguir las elevadas temperaturas y presiones necesarias, los científicos de la «General Electric Company» consiguieron al fin esta hazaña en 1955. Se lograron presiones de 100.000 atmósferas o más, con temperaturas que alcanzaban los 2.500 °C. Además, se empleó una pequeña cantidad de metal, como el cromo, para formar una película líquida a través del grafito. Fue en esta película en la que el grafito se convirtió en diamante. En 1962, se llegó a una presión de 200.000 atmósferas y una temperatura de 5.000 °C. El grafito se convirtió directamente en diamante, sin emplear un catalizador.
Los diamantes sintéticos son demasiado pequeños e impuros para emplearlos como gemas, pero en la actualidad se producen comercialmente como abrasivos y herramientas de corte, e incluso son una fuente importante para tales productos. A fines de esa década, se llegó a producir un pequeño diamante de una calidad apta en gemología.
Un producto más nuevo conseguido por la misma clase de tratamiento puede sustituir a los usos del diamante. Un compuesto de boro y nitrógeno (nitruro bórico) es muy similar en propiedades al grafito (excepto que el nitruro bórico es blanco en vez de negro). Sometido a las elevadas temperaturas y presiones que convierten al grafito en diamante, el nitruro de boro lleva a cabo una conversión similar. Desde una disposición cristalina como la del grafito, los átomos de nitruro de boro se convierten en una parecida a la del diamante. En su nueva forma se denomina borazón. El borazón es cuatro veces más duro que el carborundo. Además, posee la gran ventaja de ser más resistente al calor. A una temperatura de 900 °C el diamante arde, pero el borazón permanece incambiable.
El boro posee un electrón menos que el carbono y el nitrógeno un electrón más. Combinados ambos, alternativamente, llegan a una situación muy parecida a la de la disposición carbono-carbono, pero existe una pequeña diferencia respecto de la simetría del diamante. Por lo tanto, el boro no es tan duro como el diamante.
Naturalmente, los trabajos de Bridgman sobre las presiones elevadas no constituyen la última palabra. A principios de los años 1980, Peter M. Bell, de la «Institución Carnegie», empleó un aparato que oprime los materiales entre dos diamantes, por lo que consiguió alcanzar presiones de hasta 1.500.000 atmósferas, por encima de las dos quintas partes de las existentes en el centro de la Tierra. Creo que es posible con su instrumento llegar a los 17.000.000 de atmósferas antes de que los mismos diamantes se vean afectados.
En el Instituto de Tecnología de California, se han empleado ondas de choque para producir presiones momentáneas aún más elevadas: tal vez hasta de 75.000.000 atmósferas.
Metales
La mayor parte de los elementos de la tabla periódica son metales. En realidad, sólo 20 de los 102 pueden considerarse como no metálicos. Sin embargo, el empleo de los metales se introdujo relativamente tarde. Una de las causas es la de que, con raras excepciones, los elementos metálicos están combinados en la Naturaleza con otros elementos y no son fáciles de reconocer o extraer. El hombre primitivo empleó al principio sólo materiales que pudieran manipularse mediante tratamiento simples, como cincelar, desmenuzar, cortar y afilar. Ello limitaba a huesos, piedras y madera los materiales utilizables.
Su iniciación al uso de los metales se debió al descubrimiento de los meteoritos, o de pequeños núcleos de oro, o del cobre metálico presente en las cenizas de los fuegos hechos sobre rocas que contenían venas de cobre. En cualquier caso se trataba de gentes lo bastante curiosas (y afortunadas) como para encontrar las extrañas y nuevas sustancias y tratar de descubrir las formas de manejarlas, lo cual supuso muchas ventajas. Los metales diferían de la roca por su atractivo brillo una vez pulimentados. Podían ser golpeados hasta obtener de ellos láminas, o ser transformados en varillas. Podían ser fundidos y vertidos en un molde para solidificarlos. Eran mucho más hermosos y adaptables que la piedra, e ideales para ornamentación. Probablemente se emplearon para esto mucho antes que para otros usos.
Al ser raros, atractivos y no alterarse con el tiempo, los metales llegaron a valorarse hasta el punto de convertirse en un medio reconocido de intercambio. Al principio, las piezas de metal (oro, plata o cobre) tenían que ser pesadas por separado en las transacciones comerciales, pero hacia el 700 a. de J.C. fabricaron ya patrones de metal algunas entidades oficiales en el reino de Lidia, en Asia Menor, y en la isla egea de Egina. Aún hoy seguimos empleando las monedas.
Lo que realmente dio valor a los metales por sí mismos fue el descubrimiento de que algunos de ellos podían ser transformados en una hoja más cortante que la de la piedra. Más aún, el metal era duro. Un golpe que pudiera romper una porra de madera o mellar un hacha de piedra, sólo deformaba ligeramente un objeto metálico de tamaño similar. Estas ventajas compensaban el hecho de que el metal fuera más pesado que la piedra y más difícil de obtener.
El primer metal obtenido en cantidad razonable fue el cobre, que se usaba ya hacia el 4000 a. de J.C. Por sí solo, el cobre era demasiado blando para permitir la fabricación de armas o armaduras (si bien se empleaba para obtener bonitos ornamentos), pero a menudo se encontraba en la mena aleado con una pequeña cantidad de arsénico o antimonio, lo cual daba por resultado una sustancia más dura que el metal puro. Entonces se encontrarían algunas menas de cobre que contendrían estaño. La aleación de cobre-estaño (bronce) era ya lo suficientemente dura como para utilizarla en la obtención de armas. El hombre aprendió pronto a añadir el estaño. La Edad del Bronce remplazó a la de la Piedra, en Egipto y Asia Occidental, hacia el 3500 a. de J.C., y en el sudeste de Europa, hacia el 2000 a. de J.C. La Ilíada y La Odisea, de Hornero, conmemoran este período de la cultura.
Aunque el hierro se conoció tan pronto como el bronce, durante largo tiempo los meteoritos fueron su única fuente de obtención. Fue, pues, sólo un metal precioso, limitado a empleos ocasionales, hasta que se descubrieron métodos para fundir la mena de hierro y obtener así éste en cantidades ilimitadas. La fundición del hierro se inició, en algún lugar del Asia Menor, hacia el 1400 a. de J.C, para desarrollarse y extenderse lentamente.
Un ejército con armas de hierro podía derrotar a otro que empleara sólo las de bronce, ya que las espadas de hierro podían cortar las de bronce. Los hititas de Asia Menor fueron los primeros en utilizar masivamente armas de hierro, por lo cual vivieron un período de gran poder en el Asia Occidental. Los asirios sucedieron a los hititas. Hacia el 800 a. de J.C. tenían un ejército completamente equipado con armas de hierro, que dominaría el Asia Occidental y Egipto durante dos siglos y medio. Hacia la misma época, los dorios introdujeron la Edad del Hierro en Europa, al invadir Grecia y derrotar a los aqueos, que habían cometido el error de seguir en la Edad del Bronce.
Hierro y acero
El hierro se obtiene, esencialmente, calentando con carbón la mena de hierro (normalmente, óxido férrico). Los átomos de carbono extraen el oxígeno del óxido férrico, dejando una masa de hierro puro. En la Antigüedad, las temperaturas empleadas no fundían el hierro, por lo cual, el producto era un metal basto, que podía moldearse golpeándolo con un martillo, es decir, se obtenía el «hierro forjado». La metalurgia del hierro a gran escala comenzó en la Edad Media. Se emplearon hornos especiales y temperaturas más elevadas, que fundían el hierro. El hierro fundido podía verterse en moldes para formar coladas, por lo cual se llamó «hierro colado». Aunque mucho más barato y duro que el hierro forjado, era, sin embargo, quebradizo y no podía ser golpeado con el martillo.
La creciente demanda de ambas formas de hierro desencadenó una tala exhaustiva de los bosques ingleses, por ejemplo, pues Inglaterra consumía su madera en las fraguas. Sin embargo, allá por 1780, el herrero inglés Abraham Darby demostró que el coque (hulla carbonizada) era tan eficaz como el carbón vegetal (leña carbonizada), si no mejor. Así se alivió la presión ejercida sobre los bosques y empezó a imponerse el carbón mineral como fuente de energía, situación que duraría más de un siglo.
Finalmente, en las postrimerías del siglo XVIII, los químicos —gracias a las investigaciones del físico francés René-Antoine Ferchault de Réaumur— comprendieron que lo que determinaba la dureza y resistencia del hierro era su contenido en carbono. Para sacar el máximo partido a tales propiedades, el contenido de carbono debería oscilar entre el 0,2 y el 1,5 %; así, el acero resultante era más duro y resistente, más fuerte que el hierro colado o forjado. Pero hasta mediados del siglo XIX no fue posible mejorar la calidad del acero, salvo mediante un complicado procedimiento, consistente en agregar la cantidad adecuada de carbono al hierro forjado (labor cuyo coste era comparativamente muy elevado). Por tanto, el acero siguió siendo un metal de lujo, usado sólo cuando no era posible emplear ningún elemento sustitutivo, como en el caso de las espadas y los muelles.
La Edad del Acero la inició un ingeniero británico llamado Henry Bessemer. Interesado, al principio, ante todo, en cañones y proyectiles, Bessemer inventó un sistema que permitía que los cañones dispararan más lejos y con mayor precisión. Napoleón III se interesó en su invento y ofreció financiar sus experimentos. Pero un artillero francés hizo abortar la idea, señalando que la explosión propulsora que proyectaba Bessemer destrozaría los cañones de hierro colado que se empleaban en aquella época. Contrariado, Bessemer intentó resolver el problema mediante la obtención de un hierro más resistente. No sabía nada sobre metalurgia, por lo cual podía tratar el problema sin ideas preconcebidas. El hierro fundido era quebradizo por su contenido en carbono. Así, el problema consistía en reducir el contenido de este elemento. ¿Por qué no quemar el carbono y disiparlo, fundiendo el hierro y haciendo pasar a través de éste un chorro de aire? Parecía una idea ridícula. Lo normal era que el chorro de aire enfriase el metal fundido y provocase su solidificación. De todas formas, Bessemer lo intentó, y al hacerlo descubrió que ocurría precisamente lo contrario. A medida que el aire quemaba el carbono, la combustión desprendía calor y se elevaba la temperatura del hierro, en vez de bajar. El carbono se quemaba muy bien. Mediante los adecuados controles podía producirse acero en cantidad y a un costo comparativamente bajo.
En 1856, Bessemer anunció su «alto horno». Los fabricantes de hierro adoptaron el método con entusiasmo; pero luego lo abandonaron, al comprobar que el acero obtenido era de calidad inferior. Bessemer descubrió que la mena de hierro empleada en la industria contenía fósforo (elemento que no se encontraba en sus muestras de menas). A pesar de que explicó a los fabricantes de hierro que la causa del fracaso era el fósforo, éstos se negaron a hacer una prueba. Por consiguiente, Bessemer tuvo que pedir prestado dinero e instalar su propia acería en Sheffield. Importó de Suecia mena carente de fósforo y produjo en seguido acero a un precio muy inferior al de los demás fabricantes de hierro.
En 1875, el metalúrgico británico Sidney Gilchrist Thomas descubrió que revistiendo el interior del horno con piedra caliza y magnesio, podía extraer fácilmente el fósforo del hierro fundido. Con este sistema podía emplearse en la fabricación del acero casi cualquier tipo de mena. Mientras tanto, el inventor angloalemán Karl Wilhelm Siemens desarrolló, en 1868, el «horno de solera abierta», en el cual la fundición bruta era calentada junto con mena de hierro. Este proceso reducía también el contenido del fósforo en la mena.
Ya estaba en marcha la «Edad del Acero». El nombre no es una simple frase. Sin acero, serían casi inimaginables los rascacielos, los puentes colgantes, los grandes barcos, los ferrocarriles y muchas otras construcciones modernas, y, a pesar del reciente empleo de otros metales, el acero sigue siendo el metal preferido para muchos objetos, desde el bastidor de los automóviles hasta los cuchillos.
(Desde luego, es erróneo pensar que un solo paso adelante puede aportar cambios trascendentales en la vida de la Humanidad. El progreso ha sido siempre el resultado de numerosos adelantos relacionados entre sí, que forman un gran complejo. Por ejemplo, todo el acero fabricado en el mundo no permitiría levantar los rascacielos sin la existencia de ese artefacto cuya utilidad se da por descontada con excesiva frecuencia: el ascensor. En 1861, el inventor americano Elisha Graves Otis patentó un ascensor hidráulico, y en 1889, la empresa por él fundada instaló los primeros ascensores eléctricos en un edificio comercial neoyorquino.)
Una vez obtenido con facilidad acero barato, se pudo experimentar con la adición de otros metales («aleaciones de acero»), para ver si podía mejorarse aún más el acero. El experto en metalurgia británico Robert Abbott Hadfield fue el primero en trabajar en este terreno. En 1882 descubrió que añadiendo al acero un 13 % de manganeso, se obtenía una aleación más sólida, que podía utilizarse en la maquinaria empleada para trabajos muy duros, por ejemplo, el triturado de metales. En 1900, una aleación de acero que contenía tungsteno y cromo siguió manteniendo su dureza a altas temperaturas, incluso al rojo vivo y resultó excelente para máquinas-herramienta que hubieran de trabajar a altas velocidades. Hoy existen innumerables aceros de aleación para determinados trabajos, que incluyen, por ejemplo, molibdeno, níquel, cobalto y vanadio.
La principal dificultad que plantea el acero es su vulnerabilidad a la corrosión, proceso que devuelve el hierro al estado primitivo de mena del que proviene. Un modo de combatirlo consiste en proteger el metal pintándolo o recubriéndolo con planchas de un metal menos sensible a la corrosión, como el níquel, cromo, cadmio o estaño. Un método más eficaz aún es el de obtener una aleación que no se corroa. En 1913, el experto en metalurgia británico Harry Brearley descubrió casualmente esta aleación. Estaba investigando posibles aleaciones de acero que fueran especialmente útiles para los cañones de fusil. Entre las muestras descartó como inadecuada una aleación de níquel y cromo. Meses más tarde advirtió que dicha muestra seguía tan brillante como al principio, mientras que las demás estaban oxidadas. Así nació el «acero inoxidable». Es demasiado blando y caro para emplearlo en la construcción a gran escala, pero da excelentes resultados en cuchillería y en otros objetos donde la resistencia a la corrosión es más importante que su dureza.
Puesto que en todo el mundo se gastan algo así como mil millones de dólares al año en el casi inútil esfuerzo de preservar de la corrosión el hierro y el acero, prosiguieron los esfuerzos en la búsqueda de un anticorrosivo. En este sentido es interesante un reciente descubrimiento: el de los pertecnenatos, compuestos que contienen tecnecio y protegen el hierro contra la corrosión. Como es natural, este elemento, muy raro — fabricado por el hombre—, nunca será lo bastante corriente como para poderlo emplear a gran escala, pero constituye una valiosa herramienta de trabajo. Su radiactividad permite a los químicos seguir su destino y observar su comportamiento en la superficie del hierro. Si esta aplicación del tecnecio conduce a nuevos conocimientos que ayuden a resolver el problema de la corrosión, ya sólo este logro bastaría para amortizar en unos meses todo el dinero que se ha gastado durante los últimos 25 años en la investigación de elementos sintéticos.
Una de las propiedades más útiles del hierro es su intenso ferromagnetismo. El hierro mismo constituye un ejemplo de «imán débil». Queda fácilmente imantado bajo la influencia de un campo eléctrico o magnético, o sea, que sus dominios magnéticos (véase capítulo 4), son alineados con facilidad. También puede desimantarse muy fácilmente cuando se elimina el campo magnético, con lo cual los dominios vuelven a quedar orientados al azar. Esta rápida pérdida de magnetismo puede ser muy útil, al igual que en los electroimanes, donde el núcleo de hierro es imantado con facilidad al dar la corriente; pero debería quedar desimantado con la misma facilidad cuando cesa el paso de la corriente.
Desde la Segunda Guerra Mundial se ha conseguido desarrollar una nueva clase de imanes débiles: las «ferritas», ejemplos de las cuales son la ferrita de níquel (Fe2O4Ni) y la ferrita de manganeso (Fe2O4Mn), que se emplean en las computadoras como elementos que pueden imantadas o desimantarlas con la máxima rapidez y facilidad.
Los «imanes duros», cuyos dominios son difíciles de orientar o, una vez orientados, difíciles de desorientar, retienen esta propiedad durante un largo período de tiempo, una vez imantados. Varias aleaciones de acero son los ejemplos más comunes, a pesar de haberse encontrado imanes particularmente fuertes y potentes entre las aleaciones que contienen poco o ningún hierro. El ejemplo más conocido es el del «alnico», descubierto en 1913 y una de cuyas variedades está formada por aluminio, níquel y cobalto, más una pequeña cantidad de cobre. (El nombre de la aleación está compuesto por la primera sílaba de cada una de las sustancias.)
En la década de 1950 se desarrollaron técnicas que permitían emplear como imán el polvo de hierro; las partículas eran tan pequeñas, que consistían en dominios individuales; éstos podían ser orientados en el seno de una materia plástica fundida, que luego se podía solidificar, sin que los dominios perdieran la orientación que se les había dado. Estos «imanes plásticos» son muy fáciles de moldear, aunque pueden obtenerse también dotados de gran resistencia.
Nuevos metales
En las últimas décadas se han descubierto nuevos metales de gran utilidad, que eran prácticamente desconocidos hace un siglo o poco más y algunos de los cuales no se han desarrollado hasta nuestra generación. El ejemplo más sorprendente es el del aluminio, el más común de todos los metales (un 60 % más que el hierro). Pero es también muy difícil de extraer de sus menas. En 1825, Hans Christian Oersted —quien había descubierto la relación que existía entre electricidad y magnetismo— separó un poco de aluminio en forma impura. A partir de entonces, muchos químicos trataron, sin éxito, de purificar el metal, hasta que al fin, en 1854, el químico francés Henri-Étienne Sainte-Clair Deville, ideó un método para obtener aluminio en cantidades razonables. El aluminio es químicamente tan activo, que se vio obligado a emplear sodio metálico (elemento más activo aún) para romper la sujeción que ejercía el metal sobre los átomos vecinos. Durante un tiempo, el aluminio se vendió a centenares de dólares el kilo, lo cual lo convirtió prácticamente en un metal precioso. Napoleón III se permitió el lujo de tener una cubertería de aluminio, e hizo fabricar para su hijo un sonajero del mismo metal; en Estados Unidos, y como prueba de la gran estima de la nación hacia George Washington, su monumento fue coronado con una plancha de aluminio sólido.
En 1886, Charles Martin Hall —joven estudiante de Química del «Oberlin College»— quedó tan impresionado al oír decir a su profesor que quien descubriese un método barato para fabricar aluminio se haría inmensamente rico, que decidió intentarlo. En un laboratorio casero, instalado en su jardín, se dispuso a aplicar la teoría de Humphry Davy, según la cual el paso de una corriente eléctrica a través de metal fundido podría separar los iones metálicos y depositarlos en el cátodo. Buscando un material que pudiese disolver el aluminio, se decidió por la criolita, mineral que se encontraba en cantidades razonables sólo en Groenlandia. (Actualmente se puede conseguir criolita sintética.) Hall disolvió el óxido de aluminio en la criolita, fundió la mezcla e hizo pasar a través de la misma una corriente eléctrica. Como es natural, en el cátodo se recogió aluminio puro. Hall corrió hacia su profesor con los primeros lingotes del metal. (Hoy se conservan en la «Aluminium Company of America».)
Mientras sucedía esto, el joven químico francés Paul-Louis Toussaint Héroult, de la misma edad que Hall (veintidós años), descubrió un proceso similar en el mismo año. (Para completar la serie de coincidencias, Hall y Héroult murieron en 1914.)
El proceso Hall-Héroult convirtió el aluminio en un metal barato, a pesar de que nunca lo sería tanto como el acero, porque la mena de aluminio es menos común que la del hierro, y la electricidad (clave para la obtención de aluminio) es más cara que el carbón (clave para la del acero). De todas formas, el aluminio tiene dos grandes ventajas sobre el acero. En primer lugar, es muy liviano (pesa la tercera parte del acero). En segundo lugar, la corrosión toma en él la forma de una capa delgada y transparente, que protege las capas más profundas, sin afectar el aspecto del metal.
El aluminio puro es más bien blando, lo cual se soluciona aleándolo con otro metal. En 1906, el metalúrgico alemán Alfred Wilm obtuvo una aleación más fuerte añadiéndole un poco de cobre y una pequeñísima cantidad de magnesio. Vendió sus derechos de patente a la metalúrgica «Durener», de Alemania, compañía que dio a la aleación el nombre de «Duraluminio».
Los ingenieros comprendieron en seguida lo valioso que resultaría en la aviación un metal ligero, pero resistente. Una vez que los alemanes hubieron empleado el «Duraluminio» en los zepelines durante la Primera Guerra Mundial, y los ingleses se enteraron de su composición al analizar el material de un zepelín que se había estrellado, se extendió por todo el mundo el empleo de este nuevo metal. Debido a que el «Duraluminio» no era tan resistente a la corrosión como el aluminio, los metalúrgicos lo recubrieron con delgadas películas de aluminio puro, obteniendo así el llamado «Alelad».
Hoy existen aleaciones de aluminio que, a igualdad de pesos, son más resistentes que muchos aceros. El aluminio tiende a remplazar el acero en todos los usos en que la ligereza y la resistencia a la corrosión son más importantes que la simple dureza. Como todo el mundo sabe, hoy es de empleo universal, pues se utiliza en aviones, cohetes, trenes, automóviles, puertas, pantallas, techos, pinturas, utensilios de cocina, embalajes, etc.
Tenemos también el magnesio, metal más ligero aún que el aluminio. Se emplea principalmente en la aviación, como era de esperar. Ya en 1910, Alemania usaba aleaciones de magnesio y cinc para estos fines. Tras la Primera Guerra Mundial, se utilizaron cada vez más las aleaciones de magnesio y aluminio.
Sólo unas cuatro veces menos abundante que el aluminio, aunque químicamente más activo, el magnesio resulta más difícil de obtener a partir de las menas. Mas, por fortuna, en el océano existe una fuente muy rica del mismo. Al contrario que el aluminio o el hierro, el magnesio se halla presente en grandes cantidades en el agua de mar. El océano transporta materia disuelta, que forma hasta un 3,5 % de su masa. De este material en disolución, el 3,7 % es magnesio. Por tanto, el océano, considerado globalmente, contiene unos dos mil billones (2.000.000.000.000.000) de toneladas de magnesio, o sea, todo el que podamos emplear en un futuro indefinido.
El problema consistía en extraerlo. El método escogido fue el de bombear el agua del mar hasta grandes tanques y añadir óxido de cal (obtenido también del agua del mar, es decir, de las conchas de las ostras). El óxido de cal reacciona con el agua y el ion del magnesio para formar hidróxido de magnesio, que es insoluble y, por tanto, precipita en la disolución. El hidróxido de magnesio se convierte en cloruro de magnesio mediante un tratamiento con ácido clorhídrico, y luego se separa el magnesio del cloro por medio de una corriente eléctrica.
En enero de 1941, la «Dow Chemical Company» produjo los primeros lingotes de magnesio a partir del agua del mar, y la planta de fabricación se preparó para decuplicar la producción de magnesio durante los años de la guerra.
De hecho, cualquier elemento que pueda extraerse del agua de mar a un precio razonable, puede considerarse como de una fuente virtualmente ilimitada, ya que, después de su uso, retorna siempre al mar. Se ha calculado que si se extrajesen cada año del agua de mar 100 millones de toneladas de magnesio durante un millón de años, el contenido del océano en magnesio descendería de su porcentaje actual, que es del 0,13, al 0,12 %.
Si el acero fue el «metal milagroso» de mediados del siglo XIX, el aluminio fue el de principios de siglo XX y el magnesio el de mediados del mismo, ¿cuál será el nuevo metal milagroso? Las posibilidades son limitadas. En realidad hay sólo siete metales comunes en la corteza terrestre. Además del hierro, aluminio y magnesio, tenemos el sodio, potasio, calcio y titanio. El sodio, potasio y calcio son demasiado activos químicamente para ser empleados como metales en la construcción. (Por ejemplo, reaccionan violentamente con el agua.) Ello nos deja sólo el titanio, que es unas ocho veces menos abundante que el hierro.
El titanio posee una extraordinaria combinación de buenas cualidades: su peso resulta algo superior a la mitad del acero; es mucho más resistente que el aluminio o el acero, difícilmente afectado por la corrosión y capaz de soportar temperaturas muy elevadas. Por todas estas razones, el titanio se emplea hoy en la aviación, construcción de barcos, proyectiles teledirigidos y en todos los casos en que sus propiedades puedan encontrar un uso adecuado.
¿Por qué ha tardado tanto la Humanidad en descubrir el valor del titanio? La razón es casi la misma que puede esgrimirse para el aluminio y el magnesio. Reacciona demasiado rápidamente con otras sustancias, y en sus formas impuras —combinado con oxígeno o nitrógeno— es un metal poco atractivo, quebradizo y aparentemente inútil. Su resistencia y sus otras cualidades positivas se tienen sólo cuando es aislado en su forma verdaderamente pura (en el vacío o bajo un gas inerte). El esfuerzo de los metalúrgicos ha tenido éxito en el sentido de que 1 kg de titanio, que costaba 6 dólares en 1947, valía 3 en 1969.
No se tienen que encauzar forzosamente las investigaciones para descubrir metales «maravillosos». Los metales antiguos (y también algunos no metales) pueden llegar a ser más «milagrosos» de lo que lo son actualmente.
En el poema The Deacon's Masterpiece, de Oliver Wendell Holmes, se cuenta la historia de una «calesa» construida tan cuidadosamente, que no tenía ni un solo punto débil. Al fin, el carricoche desapareció por completo, se descompuso en polvo. Pero había durado 100 años.
La estructura atómica de los sólidos cristalinos, tanto metales como no metales, es muy parecida a la de esta imaginaria calesa. Los cristales de un metal están acribillados por cientos de fisuras y arañazos submicroscópicos. Bajo la fuerza de la presión, en uno de esos puntos débiles puede iniciarse una fractura y extenderse a través de todo el cristal. Si, al igual que la maravillosa calesa del diácono, un cristal pudiese estar construido sin puntos débiles, tendría muchísima más resistencia.
Estos cristales sin ningún punto débil toman la forma de delgados filamentos — denominados «triquitas»— sobre la superficie de los cristales. La resistencia a la elongación de las triquitas de carbono puede alcanzar hasta las 1.400 t/cm2, es decir, de 16a 17 veces más que las del acero. Si pudieran inventarse métodos para la fabricación de metal sin defecto alguno y en grandes cantidades, encontraríamos viejos metales, que podrían ser nuevos y «milagrosos». Por ejemplo, en 1968 los científicos soviéticos fabricaron un delicado cristal de tungsteno sin defecto alguno, que podía soportar una carga de 1.635 t/cm2, cantidad respetable comparada con las 213 t/cm2 de los mejores aceros. Y aunque tales fibras puras no podían adquirirse a granel, la integración de fibras nítidas con los metales serviría para reforzarlos y fortalecerlos.
También en 1968 se inventó un nuevo método para combinar metales. Los dos métodos de importancia tradicional eran, hasta entonces, la aleación —o sea, la fusión simultánea de dos o más metales, con objeto de formar una mezcla más o menos homogénea— y la galvanización, que consiste en unir sólidamente un metal con otro. (Para ello se aplica una sutil capa de metal caro a una masa voluminosa de metal barato, de forma que la superficie resulta tan bonita y anticorrosiva como la del oro, por ejemplo, pero cuyo conjunto es casi tan económico como el cobre.)
El metalúrgico americano Newell C. Cook y sus colaboradores intentaron galvanizar con silicio una superficie de platino, empleando fluorita fundida como solución para bañar el platino. Contra todos los pronósticos, no hubo galvanización. Al parecer, la fluorita fundida disolvió la fina película de oxígeno que cubre, por lo general, casi todos los metales, incluso los más resistentes, y dejó «expuesta» la superficie del platino a los átomos de silicio. Así, pues, éstos se abrieron paso por la superficie, en lugar de unirse a ella por la parte inferior de los átomos de oxígeno. El resultado fue que una fina capa externa del platino se transformó en aleación.
Siguiendo esta inesperada línea de investigación, Cook descubrió que se podían combinar muchas sustancias para formar una aleación «galvanizada» sobre metal puro (o sobre otra aleación). Llamó a este proceso «metalización», cuya utilidad demostró. Así, se fortalece excepcionalmente el cobre agregándole un 2-4 % de berilio en forma de aleación corriente. Pero se puede obtener un resultado idéntico si se «beriliza» el cobre, cuyo costo es muy inferior al que representaría el berilio puro, elemento relativamente raro. También se endurece el acero metalizado con boro. La adición de silicio, cobalto y titanio da asimismo unas propiedades útiles.
Dicho de otra forma: si los metales maravillosos no se encuentran en la Naturaleza, el ingenio humano es capaz de crearlos.