De patie
¨nt is normaal georie¨nteerd en er zijn
een kleine subgroep patie¨nten met de ziekte van
geen geheugenstoornissen. Het taalbegrip en
Creutzfeldt-Jakob (5-10%) met een sluipend begin
de taalproductie zijn normaal, maar hij spreekt
en een veel langer ziektebeloop van twee tot vijf
dysartrisch. Bij onderzoek van de oogbewegin-
jaar.
gen zijn de saccaden traag. De sensibiliteit en
reflexen zijn zonder afwijkingen, de coo
¨rdinatie
is licht atactisch. Er zijn geen paresen, de tonus
j
24.3
De meest voorkomende symptomen
is normaal. De patie
¨nt kan nauwelijks los staan.
bij de ziekte van Creutzfeldt-Jakob
Hij kan met veel moeite enkele schuifelende
pasjes maken. Tijdens de opname gaat de
Vaak (25%) worden de eerste daadwerkelijke func-
patie
¨nt verder achteruit en na een week worden
tiestoornissen (bijvoorbeeld ataxie) voorafgegaan
actiemyoclonie
¨n opgemerkt. Op een nieuwe
door een prodromale fase van enkele weken waarin
MRI van de hersenen wordt een verhoogde sig-
de patie¨nten klagen over lusteloosheid, angst, sla-
naalintensiteit gezien beiderzijds in de basale
peloosheid, vage pijn, verminderde eetlust of ge-
kernen. Het celgetal en totaaleiwit in de liquor
wichtsverlies. Bij de eerste ziekteverschijnselen
cerebrospinalis zijn normaal, maar de liquor
kunnen drie patronen worden onderscheiden, alle
blijkt immunoreactief met antilichamen tegen
optredend bij ongeveer een derde van de patie¨nten.
14-3-3 eiwit. Een EEG laat een licht vertraagd
1 Bij presentatie kunnen niet-cognitieve sympto-
achtergrondpatroon zien, zonder specifieke
men op de voorgrond staan, zoals diplopie, ata-
afwijkingen. Na sequentieanalyse van het pri-
xie of loopstoornissen.
ongen blijkt de patie
¨nt homozygoot (methioni-
2 Er kan vanaf het begin sprake zijn van verande-
ne/methionine) te zijn op codon 129.
ringen in het cognitieve functioneren of van
Op grond van deze bevindingen wordt de
psychiatrische symptomen (zie verder).
diagnose ‘ziekte van Creutzfeldt-Jakob’ gesteld.
3 Er kan vanaf het begin sprake zijn van een com-
Deze diagnose wordt met de patie
¨nt en zijn
binatie van mentale veranderingen en andere
twee dochters besproken. De motorische en
neurologische symptomen.
cognitieve functies van patie¨nt verslechteren en
hij wordt overgeplaatst naar een hospice waar
j
24.3.1
Psychiatrische symptomen
hij elf maanden na het begin van de klachten
overlijdt. Bij postmortaal neuropathologisch
Bij de mentale veranderingen staan bij de helft van
onderzoek wordt de diagnose ziekte van
de patie¨nten psychiatrische symptomen op de
Creutzfeldt-Jakob bevestigd.
voorgrond. Dit wordt onder andere geı¨llustreerd
door Bertha E., de eerste patie¨nt die Creutzfeldt in
1920 beschreef, die zichzelf verwaarloosde en dacht
dat ze door de duivel bezeten was. In het verleden
werd op grond van de waarschijnlijkheidsdiagnose
schizofrenie zelfs elektroshocktherapie toegepast
24 Ziekte van Creutzfeldt-Jakob
263
voordat de diagnose ziekte van Creutzfeldt-Jakob
j
24.3.4
Frequentie van voorkomen
werd gesteld. Soms wordt in verband met vroege
symptomen, zoals zelfverwaarlozing, apathie of
Gegevens over de frequentie waarin de verschil-
depressie, in eerste instantie een behandeling met
lende symptomen voorkomen zijn bij zeldzame
antidepressiva gestart. Agitatie, wanen of halluci-
ziekten als de ziekte van Creutzfeldt-Jakob vaak
naties als eerste verschijnsel leiden vanzelfspre-
gebaseerd op relatief kleine patie¨ntenseries, waar-
kend eerder tot verwijzing naar een psychiater dan
bij vastgelegd is welk percentage patie¨nten in de
naar een neuroloog. Toch zal, als de beginsympto-
loop van de ziekte ooit een bepaald symptoom
men aanvankelijk als functioneel-psychiatrisch
heeft vertoond. Het ‘symptomenprofiel’ wat op die
worden geı¨nterpreteerd, in de loop van enkele we-
manier ontstaat is meestal niet representatief voor
ken de organische achtergrond snel duidelijk wor-
de verschijnselen die patie¨nten in het begin van de
den door het ontstaan van andere verschijnselen.
ziekte vertonen. De frequentieverdelingen van be-
Zo verslechterde ook de toestand van Bertha E.
ginsymptomen en symptomen die zich cumulatief
volgens de beschrijving van Creutzfeldt snel. Zij
in de loop van de ziekte voordoen, zoals weerge-
raakte in coma en overleed enkele maanden na het
geven in figuur 24.1, illustreren dit principe. Bij de
begin van haar symptomen.
vergelijking van het ziektebeeld van een indivi-
duele patie¨nt met het ‘klassieke’ klinische beeld,
j
24.3.2
Fysieke problemen
moet dus rekening worden gehouden met het sta-
dium van de ziekte waarin de betreffende patie¨nt
Een globale ernstige dementie wordt voorafgegaan
verkeert.
door geı¨soleerde corticale functiestoornissen, zoals
afasie, apraxie, agrafie, agnosie en neglect.
Stoornissen in de motoriek komen voor op
j
24.4
Varianten van de ziekte van
grond van:
Creutzfeldt-Jakob
– extrapiramidale verschijnselen, zoals rigiditeit,
tremor en dystonie;
Het klinische beeld van de ziekte van Creutzfeldt-
– piramidale afwijkingen, zoals spasticiteit en pa-
Jakob is gebaseerd op de symptomen bij de spora-
rese;
dische Creutzfeldt-Jakob, maar afhankelijk van de
– cerebellaire verschijnselen, zoals dysartrie en
oorzaak van prionziekten kunnen variaties in het
ataxie.
klinisch beeld optreden.
Myoclonus, vaak toenemend onder invloed van
j
24.4.1
BSE/ziekte van Creutzfeldt-Jakob
externe stimuli, komt met het voortschrijden van
de ziekte in toenemende frequentie voor.
In Engeland zijn tot op heden circa 160 patie¨nten
Visusafwijkingen kunnen betrekking hebben op
bekend, waarbij een oorzakelijk verband met de in
dubbelzien, veranderde waarneming van kleuren,
dat land veelvoorkomende bovine spongiform ence-
gezichtsvelddefecten, wazig zien, metamorfopsie
phalopathy (BSE) zeer aannemelijk is. Deze nieuwe
en corticale blindheid.
variant van de ziekte van Creutzfeldt-Jakob komt
Ten slotte zijn ook sensibele stoornissen, her-
voor bij een populatie die met een gemiddelde
senzenuwuitval en slaapstoornissen bij de ziekte
leeftijd van ongeveer 30 jaar veel jonger is dan
van Creutzfeldt-Jakob beschreven. Uiteindelijk
patie¨nten met sporadische vormen van de ziekte
treedt een akinetisch-mutistisch toestandsbeeld
van Creutzfeldt-Jakob (65 jaar) en de gemiddelde
op.
ziekteduur is met 15 maanden bijna 3 keer zo lang.
Het ziektebeeld werd in 1996 voor het eerst be-
j
24.3.3
Therapeutische maatregelen
schreven en veroorzaakte wereldwijd grote con-
sternatie. Een epidemie met tienduizenden
Er zijn geen specifieke therapeutische maatrege-
patie¨nten werd gevreesd, maar in de jaren 2005-
len. Voor slikstoornissen kan, afhankelijk van de
2007 waren er jaarlijks 5 patie¨nten met de ziekte
wens van de patie¨nt en familie, een voedingssonde
van Creutzfeldt-Jakob, veel minder dan volgens
worden overwogen. Myocloniee¨n kan men probe-
veel prognoses werd gevreesd. Tezamen met het
ren te verminderen door toediening van valproaat
karakteristieke neuropathologische beeld, zijn
of clonazepam, waarbij clonazepam sedatie als na-
vooral de jeugdige beginleeftijd en de lange ziek-
deel heeft.
teduur opvallend. Het is nog steeds niet duidelijk
of ook de rest van het klinische beeld zo specifiek
is. Patie¨nten met de ziekte van Creutzfeldt-Jakob
Handboek dementie
geheugenstoornissen
coördinatiestoornissen
gedragsstoornissen
cognitieve stoornissen
myoclonus
bewegingsstoornissen
EEG–afwijkingen
50%
25%
0
50%
75%
100%
bij begin van CJ
in het ziektebeloop van CJ
Figuur 24.1
Frequentieverdelingen van beginsymptomen en symptomen die zich cumulatief in de loop van de ziekte voordoen.
CJ: ziekte van Creutzfeldt-Jacob. De beginsymptomen zijn in volgorde van afnemende frequentie gerangschikt. Het
‘symptoomprofiel’ bij het begin van de ziekte van Creutzfeldt-Jakob verschilt van het beeld dat in de loop van de ziekte ontstaat. Coo
¨rdinatiestoornissen bijvoorbeeld zijn in het begin het op e
éń na meest voorkomende symptoom, terwijl
later myocloniee¨n en cognitieve stoornissen meer op de voorgrond staan.
worden in het begin van de ziekte frequent naar
tonome stoornissen. Er zijn ook families beschre-
een psychiater verwezen met depressieve of psy-
ven waarbij patie¨nten zich presenteerden met een
chotische verschijnselen, die echter ook bij spora-
spastische parese. Erfelijke vormen van prionziek-
dische vormen van de ziekte van Creutzfeldt-Jakob
ten zijn extreem zeldzaam (1 per 10 miljoen per
niet zeldzaam zijn. Bovendien is deze populatie
jaar). FFI bijvoorbeeld is nog nooit in Nederland
jonger, hetgeen verwijzing naar een psychiater
beschreven, terwijl er in ons land vooralsnog
meer voor de hand liggend maakt. Voorts komen
slechts vier families bekend zijn waarbij sprake is
klachten over pijnlijke tintelingen in het begin van
van GSS of Creutzfeldt-Jakob.
de ziekte wat vaker voor. Een goede vergelijking
tussen simultaan bestudeerde series patie¨nten met
j
24.4.3
Iatrogene Creutzfeldt-Jakob
de ziekte van Creutzfeldt-Jakob en ‘klassieke’
Creutzfeldt-Jakob is tot op heden nooit gemaakt.
Iatrogene Creutzfeldt-Jakob kan ontstaan door
Het is daardoor onduidelijk in hoeverre de ge-
overdracht van abnormaal prioneiwit via stereo-
noemde kenmerken daadwerkelijk bruikbaar zijn
tactische elektroden of via postmortaal verkregen
voor de differentie¨le diagnostiek.
humaan hypofysair groeihormoon, cornea of dura
mater. Cerebellaire symptomen (ataxie en dysar-
j
24.4.2
Erfelijke vormen van prionziekten
trie) staan bij iatrogene Creutzfeldt-Jakob sterk op
de voorgrond. Cognitieve stoornissen treden pas
Bij erfelijke vormen van prionziekten vertoont het
later in het ziektebeloop op. Overdracht via cor-
klinische beeld soms sterke overeenkomst met
neaweefsel en elektroden is wereldwijd in totaal
sporadische Creutzfeldt-Jakob, maar meestal is er
slechts viermaal beschreven. Creutzfeldt-Jakob na
sprake van een langzamer progressief beloop. Soms
gebruik van humaan groeihormoon is tot op heden
staan andere verschijnselen dan dementie op de
bij circa honderd patie¨nten aannemelijk gemaakt
voorgrond, zoals bij het Gerstmann-Straüssler-
(eéń patie¨nt in Nederland), gemiddeld dertien jaar
Scheinkersyndroom (GSS) dat vooral gekarakteri-
na het gebruik van het geı¨nfecteerde materiaal.
seerd wordt door ataxie. Patie¨nten met het erfe-
Inmiddels wordt uitsluitend met behulp van re-
lijke Fatale Familiaire Insomnie (FFI) lijden aan
combinante technieken geproduceerd groeihor-
slapeloosheid, naast cognitieve, endocriene en au-
moon gebruikt. Creutzfeldt-Jakob na implantatie
24 Ziekte van Creutzfeldt-Jakob
265
van dura mater bij neurochirurgische ingrepen is
aanvullend onderzoek geen goede andere verkla-
wereldwijd bij circa 80 patie¨nten beschreven (2
ring geeft voor de symptomen, is bepaling van het
Nederlandse patie¨nten tot op heden). Incubatietij-
14-3-3-eiwit nuttig voor de diagnostiek van de
den varie¨ren van 1,5 tot 10 jaar.
ziekte van Creutzfeldt-Jakob, maar ook liquorcon-
centraties van andere hersenspecifieke eiwitten
j
24.4.4
Kuru
(tau, NSE, S100) zijn sterk afwijkend. Voor zover
bekend is er geen directe relatie tussen het prion-
Kuru in Papua Nieuw Guinea is geassocieerd met
en het 14-3-3-eiwit. Het 14-3-3-eiwit is een normaal
kannibalistische rouwrituelen. Daarbij werd uit
cellulair eiwit dat in allerlei weefsels tot expressie
respect voor de overledene vooral door vrouwen en
komt, maar normaal in slechts zeer lage, nauwe-
kinderen hersenweefsel gegeten. Het is onduide-
lijks aantoonbare concentraties in de liquor aan-
lijk of de ziekteoverdracht daadwerkelijk toege-
wezig is. Bij snel progressieve, massale neuronale
schreven moet worden aan consumptie van her-
schade zoals bij de ziekte van Creutzfeldt-Jakob, is
senweefsel of bijvoorbeeld aan infectie tijdens de
het normale evenwicht tussen aanbod en klaring
bereiding van het materiaal. Hersenweefsel werd
van dit eiwit in de liquor blijkbaar verstoord. Bij
met de hand in bamboecilinders geschept. Tijdens
patie¨nten met de ziekte van Creutzfeldt-Jakob is in
deze bereiding kan, behalve via de orale route, ook
95% van de gevallen (sensitiviteit) het 14-3-3-eiwit
overdracht hebben plaatsgevonden via dermale of
met een ‘Western blot’ in de liquor aantoonbaar. In
conjunctivale overdracht. Bij kuru worden cogni-
patie¨ntengroepen met andere aandoeningen is dit
tieve stoornissen voorafgegaan door ataxie, dysar-
bij slechts 5% het geval (foutpositieve fractie), als
trie en diplopie. In een later stadium ontstaat er
een meningo-encefalitis, recent herseninfarct of
emotionele labiliteit met soms oncontroleerbare
een hersentumor met het overige aanvullend on-
lachbuien. Patie¨nten overlijden gemiddeld een jaar
derzoek is uitgesloten. Bij een a priori kans van
na het begin van de verschijnselen. De ziekte komt
30% op de ziekte van Creutzfeldt-Jakob in een po-
niet voor bij personen die werden geboren na het
pulatie met snel progressieve cognitieve stoornis-
staken van bovenbeschreven rouwrituelen in 1956.
sen, al of niet gecombineerd met andere neurolo-
Nog steeds zijn er echter wel incidenteel nieuwe
gische symptomen, stijgt de kans op Creutzfeldt-
ziektegevallen onder personen die ruim 40 jaar
Jakob tot ongeveer 90% bij een positieve 14-3-3-test
geleden als kind blootgesteld zijn geweest aan het
en daalt zij naar ongeveer 1% bij een negatief test-
prion-agens.
resultaat. De sensitiviteit van de 14-3-3-test is bij de
ziekte van Creutzfeldt-Jakob en erfelijke vormen
van prionziekte beduidend lager, waarschijnlijk
j
24.5
Aanvullend onderzoek
samenhangend met een trager ziektebeloop als
uiting van een minder snel voortschrijdend neu-
Hematologisch of bloedchemisch routineonder-
ronenverlies.
zoek brengt bij Creutzfeldt-Jakob geen relevante
afwijkingen aan het licht, evenmin bij vele andere
j
24.5.2
EEG
neurodegeneratieve ziekten. Hetzelfde geldt voor
onderzoek van het celaantal, de eiwitconcentratie
Voor de introductie van de 14-3-3-test op liquor,
en IgG-index van de liquor cerebrospinalis. Het
nam het EEG van oudsher een belangrijke plaats in
aantal cellen is nooit verhoogd. Het totale liquor-
bij de diagnostiek van de ziekte van Creutzfeldt-
eiwit is meestal normaal, soms licht verhoogd,
Jakob, vooral omdat al het overige hulponderzoek
maar bijna nooit (minder dan 5%) hoger dan eéń
dat indertijd beschikbaar was geen diagnostische
gram per liter. Gezien de presentatie met snel in
informatie opleverde. De precieze diagnostische
ernst toenemende neurologische verschijnselen,
waarde van de periodieke, trifasische complexen
zal bij de meeste patie¨nten een lumbale punctie
bij een vertraagd achtergrondpatroon op het EEG
worden verricht. Als daarbij een celreactie of een
is echter nooit goed onderzocht. Cohort-onderzoe-
extreem hoog eiwit wordt gevonden, dan kan de
ken maken echter wel duidelijk dat de sensitiviteit
diagnose ziekte van Creutzfeldt-Jakob in principe
beperkt is tot ongeveer 60% en op grond van inci-
worden verworpen.
dentele meldingen is eveneens duidelijk dat de
genoemde afwijkingen in het EEG niet specifiek
j
24.5.1
14-3-3-test
zijn voor de ziekte van Creutzfeldt-Jakob. Groot
voordeel van EEG-onderzoek is dat het weinig in-
Bij een normaal celaantal en een eiwitconcentratie
vasief is. Bij de ziekte van Creutzfeldt-Jakob is het
lager dan eéń gram per liter, terwijl ook het overige
EEG wel vaak vertraagd maar ontbreken de trifa-
Handboek dementie
sische complexen. De kans op het vinden van EEG-
pathologisch veranderde vorm zoals die bij de
afwijkingen die de diagnose Creutzfeldt-Jakob
ziekte van Creutzfeldt-Jakob in de hersenen voor-
steunen, neemt toe met het voortschrijden van de
komt.
ziekte; het verdient daarom aanbeveling om bij
blijvende diagnostische twijfel het EEG periodiek
te herhalen.
j
24.6
Neurobiologie/neuropathologie
j
24.5.3
CT, SPECT en MR
Prioneiwit is een lichaamseigen product dat vooral
in zenuwweefsel voorkomt. Normaal prioneiwit
Afwijkingen op computertomografie (CT) van de
kan een verandering van vorm ondergaan, waar-
hersenen zijn niet specifiek (atrofie) en dragen
door pathologisch prioneiwit ontstaat dat resistent
daardoor niet bij aan de diagnose. Hetzelfde geldt
is tegen ontsmettingsprocedures. Pathologisch
voor de afwijkingen die bij single photon emission
prioneiwit kan de ziekte overdragen en de neuro-
computerized tomography (SPECT) zijn beschre-
toxiciteit van prionen (afgeleid van ‘proteinaceous
ven. De betekenis van afwijkingen bij magnetische
infectious particles’) hangt samen met deze veran-
resonantie (MR) onderzoek van de hersenen is,
deringen in ruimtelijke structuur.
voor zover onderzocht, vergelijkbaar met die van
Men neemt aan dat de pathologische vorm van
het EEG. De sensitiviteit is beperkt en de specifi-
prioneiwit soms bij toeval ontstaat, hetgeen de
citeit is vooralsnog onduidelijk, maar ook dit on-
zeer lage frequentie (0,5-1 per miljoen per jaar) van
derzoek leent zich zonder veel problemen voor
sporadische Creutzfeldt-Jakob verklaart. Als bij-
herhaling. Op T2-gewogen MR-afbeeldingen kan
voorbeeld door implantatie van geı¨nfecteerde dura
diffuus verhoogde signaalintensiteit worden ge-
mater, of door infusie van geı¨nfecteerd groeihor-
vonden in de basale kernen, thalamus en de neo-
moon, pathologisch prioneiwit in contact komt
cortex. Vergelijkbare afwijkingen in de basale
met het normaal prioneiwit in de hersenen, kan
kernen kunnen ook voorkomen bij bijvoorbeeld de
een cascade in gang gezet worden die leidt tot op-
ziekte van Leigh, de ziekte van Wilson, en hepati-
hoping van een pathologische vorm en uiteindelijk
sche encefalopathie, maar in die gevallen zijn er op
tot een iatrogene variant van de ziekte van
T1-gewogen opnamen verminderde signaalintensi-
Creutzfeldt-Jakob.
teiten in de betreffende gebieden. Bovendien zijn
deze ziekten met eenvoudig laboratoriumonder-
j
24.6.1
Kuru en de ziekte van Creutzfeldt-Jakob
zoek uit te sluiten. Mogelijk zijn de afwijkingen in
de basale kernen, thalamus en neocortex met dif-
Bij kuru en de ziekte van Creutzfeldt-Jakob neemt
fusie-gewogen MR-opnamen al in een eerder sta-
men aan dat de ziekte toegeschreven moet worden
dium detecteerbaar. Volgens recente gegevens ko-
aan ingestie van respectievelijk humaan patholo-
men afwijkingen in het pulvinar van de thalamus
gisch veranderd prioneiwit of het prioneiwit af-
bij 80% van de patie¨nten met de ziekte van
komstig van runderen. Vermoedelijk speelt ruim-
Creutzfeldt-Jakob voor.
telijke interactie tussen het normale en het patho-
logische prioneiwit hierbij een rol.
j
24.5.4
Biopt
j
24.6.2
Erfelijke vormen van prionziekten
Bij proefdieren is abnormaal prioneiwit in lymfo-
reticulair weefsel aantoonbaar. Dit heeft onder-
Erfelijke vormen van prionziekten worden veroor-
zoek naar prioneiwit in biopsiemateriaal van ton-
zaakt door mutaties in het priongen op chromo-
sillen bij patie¨nten gestimuleerd. Alleen bij
soom 20. Bij dragers van de mutatie in dergelijke
patie¨nten met de ziekte van Creutzfeldt-Jakob
families is door de veranderde aminozuursamen-
werd in tonsillen immunoreactiviteit gevonden
stelling van het prioneiwit de drempel voor con-
met antilichamen gericht tegen het prioneiwit. Bij
versie dermate verlaagd, dat er pathologisch prio-
sporadische Creutzfeldt-Jakob is dit niet het geval
neiwit in de hersenen ontstaat.
en zijn dergelijke biopten niet zinvol.
j
24.6.3
Bèta-vouwbladstructuur
j
24.5.5
Antilichamen
Analoog aan het ziekteproces bij de ziekte van
Sinds enkele jaren zijn er voor neuropathologisch
Alzheimer, ontstaan er bij prionziekten extracel-
onderzoek antilichamen beschikbaar die niet rea-
lulaire afzettingen van abnormale eiwitfibrillen in
geren met normaal prioneiwit, maar wel met de
een zogenaamde bèta-vouwbladstructuur, in dit
24 Ziekte van Creutzfeldt-Jakob
267
geval bestaande uit prioneiwit. Deze aggregaten
proberen te behandelen. Myocloniee¨n kan men
veroorzaken sponsachtige veranderingen in her-
proberen te verminderen door toediening van val-
senweefsel. Met ook astrocytaire gliose.
proaat of clonazepam, waarbij clonazepam sedatie
als nadeel heeft. Voor slikstoornissen kan, afhan-
kelijk van de wens van de patie¨nt en zijn familie,
j
24.7
Genetische aspecten
een voedingssonde worden overwogen.
Het normale priongen kent in de algemene bevol-
king een variatie op positie 129, waar het codeert
j
24.9
Preventie
voor ofwel het aminozuur methionine (M) ofwel
valine (V). Dit codon-129-polymorfisme heeft geen
De combinatie van het snelle, onvermijdelijk fatale
directe ziekteverwekkende invloed, maar beı¨n-
beloop van de ziekte van Creutzfeldt-Jakob en het
vloedt waarschijnlijk wel de gevoeligheid voor
buitenissige, deels infectieuze karakter van prio-
prionziekten. In de bevolking wordt M/M bij 37%
nen, veroorzaakt na het stellen van de diagnose
gevonden, V/V bij 12% en is ongeveer 50% hetero-
vaak onzekerheid over mogelijke besmettelijkheid
zygoot (M/V). In populaties van patie¨nten met de
voor familie en andere betrokkenen. Bij de verzor-
ziekte van Creutzfeldt-Jakob is echter slechts 16%
ging van patie¨nten met de ziekte van Creutzfeldt-
heterozygoot en 84% homozygoot. Men veronder-
Jakob en bij direct lichamelijk contact bestaat er
stelt dat dit samenhangt met het feit dat ruimte-
geen gevaar voor overdracht. Creutzfeldt-Jakob is
lijke interacties tussen prioneiwitten een belang-
niet overdraagbaar via speeksel, traanvocht, urine
rijke rol spelen bij de pathogenese: bij homozygo-
of feces. Liquor bevat echter wel infectieus materi-
ten zijn de PrP-peptiden identiek en dat zou het
aal en van bloed is dit aannemelijk. Gebruikelijke
ontstaan van prionziekten kunnen vergemakkelij-
voorzorgsmaatregelen, zoals het dragen van hand-
ken. Deze veronderstelling wordt gesteund door
schoenen en het vermijden van contact met punc-
het feit dat alle Engelse patie¨nten met de ziekte
tienaalden, worden geacht voldoende veiligheid te
van Creutzfeldt-Jakob tot nu toe homozygoot zijn
garanderen. Gebruikte materialen (naalden, gaas-
voor M op codon 129, dat bij iatrogene Creutzfeldt-
jes, buisjes) moeten behandeld worden zoals ander
Jakob homozygoten oververtegenwoordigd zijn,
besmettelijk ziekenhuisafval: verzamelen in speci-
en dat homozygote sporadische patie¨nten met
ale containers om later te worden verbrand (zie de
Creutzfeldt-Jakob een sneller ziektebeloop ken-
landelijke richtlijnen van de Werkgroep Infectie-
nen.
preventie, www.wip.nl). Bij heelkundige ingrepen
In Nederland zijn vijf stambomen bekend met
en obducties worden in die richtlijnen extra voor-
families met een autosomaal dominante vorm van
zorgen geadviseerd.
prionziekte. In deze gevallen is er sprake van ofwel
een puntmutatie ofwel een insertie in het prion-
gen. Dit biedt in principe aanknopingspunten
Literatuur
voor preklinische diagnostiek bij gezonde familie-
leden. Het zal evident zijn dat daartoe niet licht-
Collinge J, Whitfield J, McKintosh E, et al. Kuru in
vaardig overgegaan dient te worden, maar in en-
the 21st century–an acquired human prion disease
kele gevallen is dit, met goede klinisch genetische
with very long incubation periods. Lancet. 2006;
begeleiding, wel gedaan.
367:2068-74.
Collins SJ, Lawson VA, Masters CL. Transmissible
spongiform encephalopathies. Lancet. 2004;363:
j
24.8
Farmacotherapie
51-61.
Glatzel M, Stoeck K, Seeger H, et al. Human prion
Onderzoek naar medicamenteuze beı¨nvloeding
diseases: molecular and clinical aspects. Arch Neu-
van het beloop van prionziekten staat nog in de
rol. 2005;62:545-52.
kinderschoenen. In theorie en in vitro zijn er vele
Korth C, Peters PJ. Emerging pharmacotherapies for
manieren waarop de confirmatieveranderingen
Creutzfeldt-Jakob disease. Arch Neurol. 2006;63:
van het prioneiwit kunnen worden beı¨nvloed;
497-501.
vooralsnog hebben deze benaderingswijzen niet
Mendonca RA, Martins G, Lugokenski R, Rossi MD.
geleid tot een therapie die in klinisch opzicht ef-
Subacute spongiform encephalopathies. Top Magn
fectief is. Klinische trials met quinacrine (USA) en
Reson Imaging. 2005;16:213-9.
pentosan polysulfaat (UK) zijn gaande.
Pocchiari M, Puopolo M, Croes EA, et al. Predictors of
Specifieke klachten kan men symptomatisch
survival in sporadic Creutzfeldt-Jakob disease and
Handboek dementie
other human transmissible spongiform encephalo-
nar sign in variant Creutzfeldt-Jakob disease. Arch
pathies. Brain. 2004;127:2348-59.
Neurol. 2004;61:446-7.
Summers DM, Collie DA, Zeidler M et al. The pulvi-
25 Progressieve supranucleaire verlamming
W.Z. Chiu, A.J.W. Boon, J.C. van Swieten
Kernpunten
– Progressieve supranucleaire verlamming wordt klinisch gekenmerkt door een
loop- en balansstoornis waardoor patie¨nten frequent vallen en oogbewegings-
stoornissen, dysartrie, dysfagie en cognitieve stoornissen optreden.
– Progressieve supranucleaire verlamming wordt in het algemeen als een sporadi-
sche aandoening beschouwd. Maar recentelijk is familiaire aggregatie bij een klein
deel van de patie¨nten aangetoond. Het is daarom altijd zinvol te vragen naar het
voorkomen van parkinsonisme of dementie bij eerste- en tweedegraads familie-
leden.
– Op de MRI-scan kan als ondersteunende bevinding atrofie van het mesencephalon
(‘kolibri’-teken) gevonden worden en op de IBZM-SPECT-scan een verminderde
opname van het radioactief gelabeld materiaal in de basale kernen, echter door-
gaans pas in een gevorderd stadium.
– Er is voor progressieve supranucleaire verlamming geen effectieve behandeling
beschikbaar die de progressie van de ziekte kan vertragen of tegenhouden. Het
beleid is gericht op symptoombestrijding.
– Slechts bij een kwart van de patie¨nten met progressieve supranucleaire verlam-
ming wordt direct de juiste diagnose gesteld. De ziekte van Parkinson en dementie
zijn de meest gestelde foutieve diagnosen in het begin van de ziekte.
j
25.1
Inleiding
diagnose te verbeteren. Er is veel onderzoek ver-
richt, gericht op verschillen in beeldvorming en
In 1964 introduceerden Steele, Richardson en Ols-
liquoranalyse tussen progressieve supranucleaire
zewski de term Progressieve Supranucleaire Palsy
verlamming en gerelateerde aandoeningen, zoals
(PSP) ofwel Progressieve Supranucleaire Verlam-
de ziekte van Parkinson, multisysteematrofie
ming. Zij beschreven negen patie¨nten met een
(MSA), en corticobasale degeneratie (CBD). On-
verticale blikparese, pseudobulbaire verlamming,
danks deze ontwikkelingen heeft de beschrijving
axiale dystonie en dementie. Het begin van de
van Steele en collega’s niets aan waarde verloren.
ziekte verliep geleidelijk. Bij neuropathologisch
onderzoek werden neuronenverlies, gliose en
‘neurofibrillary tangles’ in de basale ganglia, de
hersenstam en het cerebellum gezien.
Er zijn internationale consensuscriteria opge-
steld om de nauwkeurigheid van de klinische

270
Handboek dementie
Casus
Een 63-jarige man kreeg geleidelijk problemen
in de vorm van vallen tijdens het fietsen. Bij het
autorijden neigde hij naar links en nam de
bochten te ruim. Daarnaast werd het hand-
schrift kleiner. Een jaar later viel het de familie
op dat hij minder aandachtig, langzamer in het
bewegen en trager tijdens gesprekken was. De
patie
¨nt veranderde van een levendige man in
iemand die alles over zich heen liet komen en
nergens meer interesse voor had. Het geheugen
bleef goed over de jaren. Het vallen begon fre-
quenter voor te komen, ook tijdens het lopen.
Hierbij was hij niet duizelig en voelde het niet
Figuur 25.1
aankomen. De spraak werd zachter en de
De zogenaamde verbaasde blik bij PSP.
patie
¨nt klaagde over stijfheid. Bij het eerste
bezoek aan een neuroloog op 65-jarige leeftijd
werden tekenen van parkinsonisme, maar geen
j
25.2
Kliniek
afwijkingen van de oogbewegingen gevonden.
De CT-scan van de hersenen was normaal.
Progressieve supranucleaire verlamming (PSP) is
Wegens parkinsonisme werd de patie
¨nt op proef
een van de meest voorkomende oorzaken van aty-
behandeld met Sinemet, echter zonder effect.
pische parkinsonismen. De prevalentie van PSP
Ruim een jaar later werden tijdens een controle
wordt geschat op 5 per 100.000, wat ongeveer 5%
trage verticale saccaden gezien. De DAT- en
van alle parkinsonismen vertegenwoordigt. De in-
IBZM-SPECT-scan waren beide afwijkend.
cidentie van PSP neemt toe met de leeftijd. Man-
Wij zagen de patie
¨nt voor het eerst op 67-
nen en vrouwen lijken even vaak aangedaan. Een
jarige leeftijd. Bij onderzoek had hij een mas-
oorzakelijke omgevingsfactor is tot op heden niet
kergelaat. De spraak was nauwelijks te verstaan.
gevonden. Opmerkelijk is een cluster van atypisch
Er was een complete verticale blikverlamming.
parkinsonisme op Guadeloupe, dat sterke gelijke-
De bewegingen in horizontaal vlak waren traag
nissen vertoont met PSP en mogelijk veroorzaakt
en niet vloeiend. De snout- en palmomentaal-
wordt door neurotoxische alkaloı¨den in lokale
reflex waren positief en de masseterreflex was
vruchten en kruidenthee.
verhoogd. Er was geen tremor. De tonus aan de
armen was verhoogd, met een tandradfeno-
j
25.2.1
Motorische verschijnselen
meen beiderzijds. Ook was er axiaal een forse
rigiditeit. De spierrekkings- en voetzoolreflexen
De symptomen beginnen geleidelijk, doorgaans
waren normaal. De patie
¨nt kon niet meer zelf-
tussen het 50e en het 70e levensjaar. Meestal is een
standig staan. Op de Mini Mental State Exami-
balansstoornis met vallen het eerste symptoom. De
nation scoorde hij 28/30 punten en op de
diagnose is niet moeilijk te stellen als andere typi-
Frontal Assesment Battery 8/18 punten. Er was
sche symptomen aanwezig zijn, zoals een verticale
een ‘applause sign’ bij de klaptest.
supranucleaire blikverlamming en een verbaasde
Wegens toegenomen zorgbehoefte werd de
blik (figuur 25.1).
patie
¨nt een halfjaar later opgenomen in het
Patie¨nten presenteren zich echter vaak met atypi-
verpleeghuis. Hij had veel last van sputum, dat
sche symptomen, wat het stellen van de diagnose
zeer moeilijk kon worden opgehoest en wegge-
in de beginfase van de ziekte lastig maakt.
slikt. Een maand later ontwikkelde hij een
Internationale consensuscriteria zijn voorhan-
pneumonie. Hij ging langzaam achteruit en is
den en maken onderscheid tussen mogelijke,
overleden. De diagnose werd post mortem
waarschijnlijke en zekere PSP (zie tabel 25.1). Voor
bevestigd.
zekere PSP is neuropathologische bevestiging ver-
eist. Deze criteria richten zich op twee belangrijke
symptomen van de ziekte: een vroeg in het ziekte-
beeld optredende balansstoornis met vallen en een
verticale supranucleaire blikverlamming. Het val-
25 Progressieve supranucleaire verlamming
271
Tabel 25.1
Internationale consensuscriteria voor PSP.
mogelijke PSP
– geleidelijke progressieve aandoening met een beginleeftijd van 40 jaar en ouder;
– vertraagde verticale saccaden en houdingsinstabiliteit met vallen in het eerste
jaar van de ziekte, of een verticale blikparese;
– geen aanwijzingen voor andere aandoeningen die bovenstaande kenmerken
kunnen verklaren.
waarschijnlijke PSP
– geleidelijke progressieve aandoening met een beginleeftijd van 40 jaar of ouder;
– verticale blikparese en houdingsinstabiliteit met vallen in het eerste jaar van de
ziekte;
– geen aanwijzingen voor andere aandoeningen die bovenstaande kenmerken
kunnen verklaren.
zekere PSP
– ‘mogelijke’ PSP of ‘waarschijnlijke’ PSP met neuropathologische bevestiging.
ondersteunende criteria
– symmetrische akinesie of rigiditeit, proximaal meer dan distaal;
– abnormale stand van de nek, vooral retrocollis;
– geringe of afwezige respons op behandeling met levodopa (L-dopa);
– vroegtijdige dysfagie en dysartrie;
– vroegtijdig begin van cognitieve achteruitgang met minstens twee van de vol-
gende kenmerken: apathie, achteruitgang in abstract denken, afgenomen ver-
bale capaciteit (verbal fluency), utilisatie- of imitatiegedrag, frontale sympto-
men.
PSP: progressieve supranucleaire parese.
len gebeurt doorgaans plots, zonder aankondiging
ten is er sprake van een onduidelijke en trage
en vaak achterover. Patie¨nten corrigeren zich niet
spraak met een spastisch karakter, wat uiteindelijk
tijdens het vallen, wat kan leiden tot ernstige ver-
kan leiden tot een complete anartrie. Andere
wondingen. De frequentie kan oplopen tot meer-
patie¨nten ontwikkelen een hypofone, monotone
dere keren per dag. In de beginfase kunnen andere
dysartrie, zoals gezien kan worden bij de ziekte
motorische symptomen ontbreken.
van Parkinson. De latentietijd tot het begin van
dysartrie en dysfagie is bij PSP-patie¨nten kort (res-
j
25.2.2
Verticale blikverlamming
pectievelijk 2 en 3,2 jaar) en verschilt duidelijk met
de ziekte van Parkinson (respectievelijk 7 en 10,8
Een verticale blikverlamming komt gemiddeld voor
jaar). Het verslikken gebeurt in eerste instantie bij
na 3 tot 4 jaar. Het onderzoek van de gladde volg-
drinken en later ook bij vast voedsel. Wanneer zich
bewegingen kan dan ook normaal zijn in het begin
een ernstige dysfagie ontwikkelt, kan plaatsing
van de ziekte. Echter de snelle saccaden zijn vaak
van een PEG-katheter noodzakelijk zijn.
eerder gestoord en kunnen vroeg in het ziektebe-
loop wel afwijkingen vertonen. De saccaden kun-
j
25.2.4
Spierstoornissen
nen hypometrisch en vertraagd zijn wanneer ge-
vraagd wordt op commando snel van het ene stati-
Dystone kenmerken ontwikkelen zich frequent,
onaire punt naar het andere te kijken. Wazig zien,
waaronder blefarospasme, axiale dystonie en een
lichtintolerantie, of dubbelzien kan aanleiding zijn
retrocollis. Er wordt verondersteld dat focale dys-
voor een initie¨le verwijzing naar een oogarts.
tonie van gezichtsspieren de oorzaak is van de ge-
fixeerde gezichtsuitdrukking bij PSP-patie¨nten.
j
25.2.3
Spraak- en slikstoornissen
Dit wordt het ‘procerus’-teken genoemd. Tremor
wordt in enkele gevallen gezien, maar doorgaans
De dysartrie neemt tijdens het beloop van de ziekte
niet de klassieke ‘geldtel’-tremor die typisch is voor
geleidelijk in ernst toe. Bij een deel van de patie¨n-
de ziekte van Parkinson. Cognitieve achteruitgang

272
Handboek dementie
en persoonlijkheidverandering treden meestal in
het beloop van de ziekte op, maar kunnen in som-
mige gevallen aan de motorische verschijnselen
voorafgaan.
j
25.2.5
Fenotypen
Een grote serie onderzoeken met pathologisch be-
vestigde PSP-patie¨nten stelde twee klinische feno-
typen vast:
– typische PSP met vallen, blikverlamming en
cognitieve disfunctie, zoals oorspronkelijk be-
schreven door Steele en collega’s;
– een asymmetrisch begin, tremor, levodopares-
pons en een langere ziekteduur. De klinische
diagnose van deze tweede groep was de ziekte
van Parkinson, zelfs bij het laatste neurologisch
bezoek voor het overlijden.
j
25.3
Diagnostiek en differentie
¨le
diagnose
De ziekte van Parkinson, MSA, CBD, frontotem-
porale dementie (FTD), Lewy-body-dementie (DLB)
en cerebrovasculaire ziekte kunnen een op PSP ge-
lijkend fenotype hebben. Een opwaartse verticale
blikverlamming wordt in enkele gevallen gevon-
den bij andere neurodegeneratieve aandoeningen
of zelfs bij gezonde ouderen. Derhalve is de ver-
onderstelling dat een neerwaartse blikverlamming
Figuur 25.2
specifieker is voor PSP. Hoewel atypische parkin-
Midsagittale T1-gewogen MRI-afbeelding met het ‘pin-
sonismen veel overlap vertonen, zijn er enkele kli-
guı¨n’- of ‘kolibrie’-teken.
nische kenmerken die kunnen helpen differentie¨-
ren. De aanwezigheid van asymmetrisch parkin-
sonisme, ernstige apraxie en ‘alien limb’-fenomeen
teristiek beeld, genaamd het ‘pinguı¨n’- of ‘koli-
is karakteristiek voor CBD, terwijl vroegtijdig en
brie’-teken (figuur 25.2 en 25.3). De sensitiviteit
ernstig autonoom disfunctioneren en cerebellaire
van atrofie van het mesencephalon vroeg in het
symptomen meestal worden gezien bij patie¨nten
beloop van de ziekte is echter onbekend, omdat de
met MSA. Bij FTD staan gedragsveranderingen op
meeste patie¨nten die zijn onderzocht in een later
de voorgrond en als er zich al parkinsonisme ont-
stadium van de ziekte verkeerden. Enkele studies
wikkelt, is het meestal in gevorderde stadia van de
toonden overlap aan van mesencephalonatrofie
ziekte. Het cognitieve verval bij DLB heeft een
tussen parkinsonismesyndromen, wat het gebruik
fluctuerend karakter met frequent visuele halluci-
in de alledaagse praktijk moeilijk maakt. Voor het
naties. Als er op de MRI vasculaire laesies in de
onderscheid met de ziekte van Parkinson kan mo-
basale ganglia, thalamus en mesencephalon wor-
gelijk een diffusiegewogen MRI zinvol zijn, aan-
den gezien, kan de diagnose ‘vasculaire PSP’ wor-
gezien een verhoogde diffusiecoe¨fficie¨nt in het
den overwogen.
putamen bij PSP-patie¨nten is gevonden. Studies
met een groter aantal patie¨nten vroeg in het ziek-
tebeloop zijn echter noodzakelijk om de werkelijke
j
25.4
Aanvullend onderzoek
diagnostische waarde hiervan te bepalen. Bij PSP,
maar ook bij de ziekte van Parkinson en MSA, kan
MRI-onderzoek van de hersenen kan atrofie van
een verminderde binding van de presynaptische
het mesencephalon laten zien. Atrofie van het me-
dopaminetransporter (DAT) worden gevonden.
sencephalon toont op sagittale coupes een karak-
Hierdoor is dit weinig behulpzaam in het onder-

25 Progressieve supranucleaire verlamming
273
ling onderscheiden van de aandoeningen. Een
verlaagde binding op een IBZM-SPECT- scan, wat
een degeneratie van postsynaptische D2-receptoren
weerspiegelt, kan vooral in de beginfase waardevol
zijn bij het onderscheid met de ziekte van Parkin-
son, ofschoon een normale IBZM-scan een PSP niet
uitsluit. Hypometabolisme van de mediofrontale
regio op een FDG-PET-scan zou PSP van de ziekte
van Parkinson, MSA en CBD kunnen differentie¨-
ren, maar de series tot nu toe zijn te klein om
definitieve conclusies te trekken. Liquoranalyse
heeft tot op heden nog geen bruikbare biomarkers
opgeleverd.
j
25.4.1
Neuropsychologie
Een subcorticale dementie met mentale traagheid
is karakteristiek voor PSP. De ernst van de cogni-
tieve stoornis kan echter varie¨ren tussen PSP-
patie¨nten. Apathie, initiatiefverlies en mentale
traagheid worden regelmatig gezien en zijn waar-
Figuur 25.3
schijnlijk het gevolg van een disfunctie van de or-
Axiale T2-gewogen MRI-afbeelding van het mesence-
bito- en mediofrontale circuits. De emotionele uit-
phalon met een diameter van 12,1 mm.
ing kan veranderen, varie¨rend van ‘emotionele in-
continentie’ tot emotionele afstomping. Bij de
meerderheid ontwikkelt zich een executieve dis-
j
25.5
Neuropathologische veranderingen
functie met verminderde verbale fluency, verstoord
bij PSP
abstract denken, moeite met plannen en set shifting.
Statistische analyse van klinische symptomen in de
PSP wordt gezien als een ‘tauopathie’, gekarakte-
eerste 2 jaar in een groep van 150 PSP-patie¨nten
riseerd door aggregaten van gehyperfosforyleerd
heeft aangetoond dat er een frontale presentatie
tauproteı¨ne. Tauproteı¨ne bindt zich aan microtu-
met een cluster van cognitieve disfunctie en ge-
bulen die belangrijk zijn voor de stabiliteit van het
dragsveranderingen bij ongeveer 20% van de
neuronale cytoskelet. Andere tauopathiee¨n zijn
patie¨nten bestaat.
CBD, FTD met taupathologie met of zonder tau-
De Frontal Assessment Battery (FAB), een korte
mutaties en de ziekte van Alzheimer.
test om het disexecutief syndroom te beoordelen,
Macroscopisch onderzoek van de hersenen bij
kan behulpzaam zijn. Een waarde van onder de 15,
PSP-patie¨nten vertoont doorgaans atrofie van het
wijzend op een hoog vermoeden van betrokken-
mesencephalon en depigmentatie van de substan-
heid van de frontaalkwab, komt veelvuldig voor bij
tia nigra en in een enkel geval tevens milde atrofie
PSP-patie¨nten. Vooral de verbale fluency is aange-
van de frontaalkwabben. Neuronenverlies, gliose,
daan bij PSP-patie¨nten. De Mini Mental State
neurofibrillaire tangles (NFT’s), coiled bodies (CB’s)
Examination (MMSE) is over het algemeen slechts
en neuropil threads (NT’s) worden gezien in de basale
mild afwijkend met een gemiddelde score van 24.
ganglia en hersenstam bij microscopisch onder-
Een andere differentie¨rende test is de klaptest, die
zoek. De aanwezigheid van taupositieve tufted as-
de motorische controle beoordeelt. Patie¨nten wor-
trocyten wordt beschouwd als zeer specifiek voor
den gevraagd de onderzoeker na te doen en zo snel
PSP (figuur 25.4 en 25.5). De neocortex, in het bij-
mogelijk driemaal in hun handen te klappen. PSP-
zonder de motorische cortex, is in sommige geval-
patie¨nten hebben de neiging om vaker dan drie
len betrokken en de ernst ervan wordt geassocieerd
keer te klappen en kunnen in sommige gevallen
met de cognitieve stoornis. Het ruggenmerg is nog
niet stoppen (‘applause sign’). Dit weerspiegelt de
niet routinematig bij PSP onderzocht.
combinatie van frontale disfunctie (motorische
planning) en pathologie van de basale ganglia (on-
vermogen om een automatische activiteit te stop-
pen).
274
Handboek dementie
Figuur 25.4
Figuur 25.5
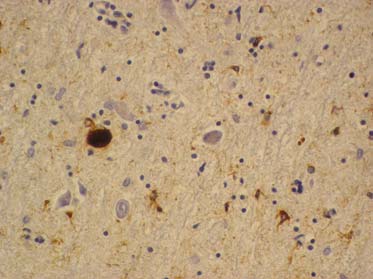
Rondvormige neurofibrillaire tangle.
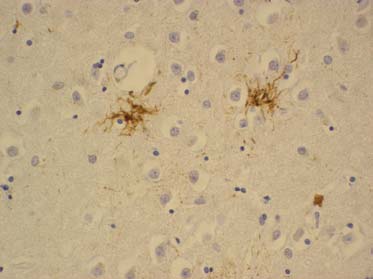
Zogenaamde tufted astrocyten (spinvormige structuren).
j
25.6
Genetische aspecten
sie van de ziekte kan vertragen of tegenhouden.
Het beleid is tot nu toe vooral gericht op symp-
De tau-aggregaten in PSP-hersenen zijn waar-
toombestrijding. Zeer recentelijk is een hoopge-
schijnlijk van etiologisch belang. PSP is in sterke
vende gerandomiseerde klinische trial met Co-en-
mate geassocieerd met een specifiek haplotype
zym Q10 gepubliceerd door Stamelou en collega’s.
(aangeduid als H1) rond het tau-gen. PSP- (en ook
Co-enzym Q10 lijkt op korte termijn klinisch een
CBD-) patie¨nten vertonen significant vaker het H1
milde verbetering te geven en de cerebrale ener-
haplotype en het H1/H1 genotype. Dit haplotype en
giehuishouding te verbeteren. Er zal echter meer
genotype kunnen derhalve als risicofactoren voor
onderzoek gedaan moeten worden om deze resul-
PSP worden beschouwd. Een van de hypothesen is
taten te bevestigen en het langetermijneffect in
dat het H2 haplotype mogelijk beschermt tegen
kaart te brengen. Levodopa laat een bescheiden en
het ontstaan van de ziekte. Om andere loci van het
geleidelijke respons bij een kwart van de patie¨nten
betrokken genoom te identificeren, is er recentelijk
zien. Toch kan het voorschrijven van levodopa in
een genoomwijde associatiestudie uitgevoerd. Of-
een vroeg stadium van nut zijn, zowel therapeu-
schoon PSP in het algemeen als een sporadische
tisch gezien, als zeker ook vanuit een diagnostisch
aandoening wordt beschouwd, is een familiaire
oogpunt. Amitriptyline kan een gunstig effect
vorm de laatste jaren in diverse studies gerappor-
hebben op motorische en pseudobulbaire sympto-
teerd. Een recente case control studie laat een ver-
men, zoals slikstoornissen, dwanglachen en
hoogde kans op parkinsonisme bij eerstegraads fa-
dwanghuilen. Bij hogere doseringen (boven 50 mg/
milieleden zien, waaronder in ongeveer 7% met een
dag) is alertheid voor het optreden van bijwerkin-
autosomaal dominant overervingpatroon. Eerder
gen geboden. Ernstige dystonie kan worden be-
zijn reeds enkele families met PSP als gevolg van
handeld met injecties met botulinetoxine. Vooral
mutaties in het MAPT-gen beschreven.
langdurige behandeling van blefarospasme met
botulinetoxine is bewezen effectief. Er is tevens
een gunstig effect gemeld bij retrocollis en orofa-
j
25.7
Prognose en beleid
ciale dystonie. Fysio- of oefentherapie kan in com-
binatie met loophulpmiddelen, zoals een ver-
PSP is een invaliderende aandoening met een ge-
zwaarde wandelstok of rollator, nuttig zijn ter
middelde periode van vijf jaar tussen ziekteaan-
verbetering van de loop- en balansstoornis. Ook
vang en het rolstoelafhankelijke stadium. De
het dragen van schoeisel met hakken kan soms het
spraak wordt na gemiddeld zes jaar onverstaan-
achterovervallen voorkomen. Logopedie kan zin-
baar. Het vroegtijdig voorkomen van pseudobul-
vol zijn bij dysartrie en dysfagie. In een later sta-
baire problemen, vallen, dubbelzien en hogere
dium kan plaatsing van een PEG-katheter nood-
leeftijd bij de ziekteaanvang hebben een verhoogd
zakelijk zijn. Faciliteiten rond het huis zijn door-
mortaliteitsrisico. Tot op heden is er voor PSP geen
gaans noodzakelijk, zodat patie¨nten zo lang mo-
effectieve behandeling beschikbaar die de progres-
gelijk bij hun verwanten kunnen verblijven.
25 Progressieve supranucleaire verlamming
275
Omdat de ziekte progressief en invaliderend is, is
Litvan I, Hauw JJ, Bartko JJ, et al. Validity and relia-
de belasting voor verzorgenden aanzienlijk. Ade-
bility of the preliminary NINDS neuropathologic
quaat vervolgen vanaf het begin van de ziekte is
criteria for progressive supranuclear palsy and rela-
derhalve belangrijk. In Nederland richt de Parkin-
ted disorders. J Neuropathol Exp Neurol. 1996;55(1):
son Vereniging zich ook op mensen met PSP en
97-105.
hun naasten, onder andere door het geven van
Rizzo G, Martinelli P, Manners D, et al. Diffusion-
voorlichting.
weighted brain imaging study of patients with
Ondanks het feit dat er veel inzicht is verkregen
clinical diagnosis of corticobasal degeneration, pro-
in verschillende aspecten van de ziekte, is er dus
gressive supranuclear palsy and Parkinson’s disease.
nog geen behandeling voor de ziekte. Verder on-
Brain. 2008;131:2690-700.
derzoek is dan ook noodzakelijk om het pathofy-
Schrag A, Ben-Shlomo Y, Quinn NP. Prevalence of
siologische proces van deze ernstige ziekte verder
progressive supranuclear palsy and multiple system
te belichten. Met deze kennis zou de ontwikkeling
atrophy: a cross-sectional study. Lancet. 1999;
van therapeutische interventies mogelijk kunnen
354(9192):1771-5.
worden.
Stamelou M, Reuss A, Pilatus U, et al. Short-term
effects of coenzyme Q10 in progressive supranuclear
palsy: a randomized, placebo-controlled trial. Mov
Literatuur
Disord 2008;23(7):942-9.
Steele JC, Richardson JC, Olszewski J. Progressive
Donker Kaat L, Boon AJW, Azmani A, et al. Familial
supranuclear palsy. A heterogeneous degeneration
aggregation of parkinsonism in Progressive supra-
involving the brain stem, basal ganglia and cerebe-
nuclear palsy Neurology. Accepted for publication.
llum with vertical gaze and pseudobulbar palsy,
Donker Kaat L, Boon AJ, Kamphorst W, et al. Frontal
nuchal dystonia and dementia. Arch Neurol. 1964;
presentation in progressive supranuclear palsy.
10:333-59.
Neurology. 2007;69(8):723-9.
Williams DR, de Silva R, Paviour DC, et al. Characte-
Litvan I, Agid Y, Calne D, et al. Clinical research cri-
ristics of two distinct clinical phenotypes in patho-
teria for the diagnosis of progressive supranuclear
logically proven progressive supranuclear palsy:
palsy (Steele-Richardson-Olszewski syndrome):
Richardson’s syndrome and PSP-parkinsonism.
report of the NINDS-SPSP international workshop.
Brain 2005;128(Pt 6):1247-58.
Neurology. 1996;47(1):1-9.
26 Corticobasale degeneratie
J.J. de Vries, K.L. Leenders
Kernpunten
– Corticobasale degeneratie is een zeldzame neurodegeneratieve aandoening
gekenmerkt door een opmerkelijk asymmetrisch akinetisch-rigide syndroom met
hogere corticale functiestoornissen (bijvoorbeeld apraxie, dysfasie, agrafesthesie).
– Verschillende nosologische entiteiten kunnen aanleiding geven tot een cortico-
basaal syndroom.
– Opvallend asymmetrische cerebrale atrofie en hypometabolisme contralateraal
aan de klinisch meest aangedane zijde en asymmetrische vertraging van het EEG-
patroon kan de diagnose ondersteunen.
– Een curatieve behandeling ontbreekt en ook effectieve, symptomatische behan-
delingen zijn niet of nauwelijks voor CBD voorhanden.
j
26.1
Inleiding
stoornissenkliniek toont een frequentie van 0,4%
(30/7500 patie¨nten). Mannen zijn mogelijk iets
Corticobasale degeneratie (CBD) is een zeldzame
meer aangedaan dan vrouwen, hoewel anderen te-
neurodegeneratieve aandoening waarbij zowel ce-
genovergestelde resultaten vinden. De incidentie is
rebrale cortices als basale ganglia zijn aangedaan.
geschat op <1 per 100.000 per jaar.
Van oudsher wordt CBD binnen het kader van de
De (typische) klinische verschijnselen, aanvul-
parkinsonistische beelden als een aparte nosologi-
lende diagnostiek, pathofysiologie, differentie¨le
sche entiteit beschouwd. De heterogene presenta-
diagnose en behandeling zullen achtereenvolgens
tie, vooral in de beginstadia, de overlap met andere
in dit hoofdstuk worden besproken.
neurodegeneratieve aandoeningen als progressieve
supranucleaire blikverlamming (PSP) of de ziekte
van Pick en de daarbij behorende pathosfysiologi-
Casus
sche mechanismen, doen de vraag rijzen of er niet
eerder sprake zou kunnen zijn van een ‘corticoba-
Een 72-jarige man meldt zich voor het eerst op
saal syndroom’ (CBS).
de polikliniek neurologie met spraakproblemen.
Sinds de eerste beschrijving van het syndroom
Hierbij is hij niet langer in staat om zijn
door Rebeiz en collega’s in de jaren zestig van de
gedachten op vloeiende wijze uit te spreken.
vorige eeuw zijn er vele studies gedaan naar de
Gaandeweg blijkt de spraakstoornis progressief,
klinische presentatie, pathologie en diagnostiek.
waarbij ook het begrip vermindert en hij niet
Tot op dit moment zijn er geen echter geen goede
langer in staat is te schrijven omdat de rechter-
epidemiologische gegevens voor CBD voorhanden.
arm ‘onhandig’ zou zijn. Deze onhandigheid
Een databasestudie van een Italiaanse bewegings-
Handboek dementie
niet altijd of pas laat in het ziektebeloop gevonden.
beperkt zich niet tot de arm, maar zit ook in de
Het ‘alien limb’-fenomeen kan worden gezien als
benen en leidt tot een vreemde loopstoornis. Bij
een zuiver motorisch dan wel sensorisch ver-
onderzoek wordt vervolgens gevonden:
schijnsel, welke niet eerder bij een andere vorm
– expressieve afasie (soms ook receptief);
van parkinsonisme is beschreven. Het fenomeen
– onvermogen blik gericht op een punt te
wordt afwisselend gedomineerd door dispraktische
houden;
eigenschappen of door frontaalkwabdisfunctie.
– levitatie van de rechterarm bij lopen;
– gestoorde corticale sensibiliteit (cijferschrij-
j
26.2.2
Hogere cerebrale functiestoornissen
ven);
– constructieve en ideomotorische apraxie (de
Hogere cerebrale functiestoornissen zijn onder an-
patie
¨nt steekt de vinger in de keel als hij
dere corticale sensibele stoornissen, dementie,
‘tandenpoetsen’ moet uitbeelden);
apraxie, frontale ontremming en dysfasie. De
– gestoord lichaamsschema.
meeste CBD-patie¨nten zijn zich niet bewust van
afwijkingen in de sensoriek of hebben slechts een
Beeldvormend en nucleair geneeskundig
‘vreemd doof’ gevoel in het aangedane ledemaat.
onderzoek bevestigen de klinische diagnose
Bij het neurologisch onderzoek worden doorgaans
corticobasale degeneratie: er is unilateraal
bij testen van de elementaire sensibiliteit (aanra-
hemisferale atrofie, hypometabolisme van de
kings- en pijnzin) geen afwijkingen gevonden. Er
frontoparie
¨tale cortex en striatum contralate-
is wel een duidelijk verminderde bewegingszin,
raal aan de klinisch meest aangedane zijde.
tweepuntsdiscriminatie, stereognosis, grafesthesie
(cijferschrijven) en aanwijzingen voor tactiele ex-
tinctie. Net als het ‘alien limb’-fenomeen worden
deze corticaal sensibele stoornissen niet bij de
j
26.2
Kliniek
ziekte van Parkinson of PSP gezien.
Een voor CBD klassieke, klinische presentatie is
j
26.2.3
Apraxie
een langzaam progressief en opmerkelijk asym-
metrisch akinetisch-rigide en dystoon beeld, bij
Een ander belangrijk en vaak vroeg in de ziekte
een persoon van gemiddeld 60 jaar met hogere
optredend symptoom is apraxie. Dit betreft dan
cerebrale functiestoornissen. CBD debuteert echter
zowel ideationele (het uitvoeren van een sequen-
niet altijd met een progressief parkinsonisme,
tie¨le taak) als ideomotorische apraxie (het op com-
maar kan ook beginnen met een spraak- of ge-
mando uitvoeren van een beweging). Afhankelijk
dragsstoornis.
van de onderliggende etiologie is ideomotorische
apraxie bij CBD-patie¨nten gemakkelijk te testen
j
26.2.1
Bewegingsstoornissen
door te vragen te hoesten of te salueren. Bij vroeg-
tijdig ontstane dysfasie, is er meer moeite om op
De meest voorkomende bewegingsstoornissen zijn
commando te hoesten dan te salueren.
akinesie, rigiditeit, posturele instabiliteit, dysto-
Het is opvallend dat veel van de hogere corticale
nie, corticale myoclonus en een posturele/intentie
functiestoornissen terug te voeren zijn op een
tremor. In een grote serie klinisch gediagnosti-
stoornis van de parie¨tale dan wel laterofrontale
ceerde CBD-patie¨nten werden de volgende symp-
cortex. Vanuit dezelfde regio kunnen de bij CBD-
tomen gevonden: parkinsonisme (100%), hogere
patie¨nten gevonden oculomotorische stoornissen
corticale functiestoornissen (93%), apraxie (82%),
ook worden verklaard. In het begin zijn patie¨nten
loopstoornis (80%), dystonie (71%), tremor (55%),
verminderd in staat om op commando te blikken
myoclonus (55%), ‘alien limb’(42%). De laatstge-
(frontaal blikcentrum), zowel in het horizontale als
noemde spreekt tot de verbeelding doordat door-
verticale vlak, en later is er ook een probleem met
gaans een arm spontaan omhoog zweeft (‘levita-
volgen (parie¨taal oogcentrum). Ook in een vroeg
tion’) of meer gedifferentieerde bewegingen ver-
stadium kunnen oogvolgbewegingsstoornissen
toont, zoals het ongewild grijpen van een voorbij-
met een optokinetische nystagmustrommel wor-
ganger. Patie¨nten zijn zich helemaal niet bewust
den aangetoond.
van deze bewegingen. In volledige verbazing kan
Om de diagnose CBD te faciliteren hebben Boeve
de beweging worden onderdrukt of in bedwang
en collega’s klinische criteria voorgesteld (zie tabel
worden gehouden door op de ledemaat te gaan
26.2). Ook met deze criteria wordt een beperkte
zitten. Een dergelijk typische presentatie wordt
accuratesse bereikt. In een klinisch-pathologische
279
Tabel 26.1
Klinische kenmerken van Corticobasale degeneratie.
bewegingsstoornissen
hogere cerebrale functiestoornissen
andere kenmerken
parkinsonisme
corticale sensibele stoornis
oogbewegingsstoornissen
– bradykinesie
– astereognosie
– supranucleaire blikverlamming
– rigiditeit
– agrafesthesie
– ‘pursuit’
– posturele instabiliteit
dystonie
dementie
focale reflexmyoclonus
tremor
apraxie
piramidale stoornissen
– actie
– ideomotor
– hyperreflexie
– houding
– ideationeel
– babinskisymptoom
orobuccale dyskinesiee
¨n
frontale ontremming
dysfagie
‘alien limb’-fenomeen
dysfasie
– niet-vloeiend (broca-afasie)
Tabel 26.2
Criteria voor de diagnose corticobasaal syndroom volgens Boeve en collega’s.
vereiste
– geleidelijk begin en progressief beloop
kenmerken
– geen aantoonbare oorzaak (bijvoorbeeld tumor of infarct)
– corticale disfunctie zich uitend in ten minste een van de volgende kenmerken:
.
asymmetrische ideomotore apraxie;
.
‘alien limb’ fenomeen;
.
corticaal sensibele stoornis;
.
visueel of sensibel hemineglect;
.
constructionele apraxie;
.
focale of asymmetrische myoclonus;
.
apractische spraak of niet-vloeiende afasie.
– extrapiramidale disfunctie zich uitend in ten minste een van de volgende symp-
tomen:
.
focale of asymmetrische rigiditeit zonder een duidelijke en voortdurende
L-doparespons;
.
focale of asymmetrische dystonie.
ondersteunende
– focale of gelateraliseerde cognitieve disfunctie met relatieve sparing van het
kenmerken
geheugen;
– focale of asymmetrische atrofie op CT of MRI van het cerebrum, met een
typische voorkeur voor de parie¨tofrontale cortex;
– focale of asymmeterische hypoperfusie op SPECT of PET van het cerebrum
maximaal in de parie
¨tofrontale cortex, basale ganglia en thalamus.
CT: computerized tomography; MRI: magnetic resonance imaging; SPECT: single photon emission; PET: positron emission tomography.
Handboek dementie
Tabel 26.3
Aanwijzingen voor de praktijk.
– er bestaat weinig klinische twijfel bij een patie¨nt van 60 jaar of ouder met een apert asymmetrisch akinetisch rigidesyndroom, corticale sensibele stoornis (astereognosie/agrafesthesie), apraxie en een alien-limbfenomeen;
– in het beginstadium van de ziekte is de differentie¨le diagnose met andere neurodegeneratieve aandoeningen lastig, waarbij een FDG-PET, EEG en liquoronderzoek met een behoorlijke accuratesse onderscheid kunnen aanbrengen;
– bij gevorderde ziekte kan een MRI-cerebrum asymmetrische atrofie van de hemisfeer contralateraal aan de klinisch meest aangedane lichaamshelft worden gevonden.
FDG-PET: fluorodeoxy-glucose positron emission tomography; EEG: elektro-encefalogram; MRI: magnetic resonance imaging.
studie van Josephs en collega’s is onlangs aange-
biomarkers voor CBD. Aanvullend onderzoek in de
toond dat patie¨nten met een klinische diagnose
vorm van beeldvorming of klinische neurofysiolo-
CBD vaak (50%) een ander pathologische diagnose
gie kan tijdens het leven enige ondersteuning bie-
blijken te hebben.
den.
j
26.4.1
Beeldvormend onderzoek
j
26.3
Diagnostiek en differentie
¨le
diagnose
Met de komst van ‘computerized tomography’ (CT)
en ‘magnetic resonance imaging’ (MRI) is aange-
Verschillende nosologische entiteiten kunnen lei-
toond dat er bij het vorderen van het ziekteproces
den tot het CBS. Afhankelijk van het presenterende
een asymmetrische verdeling van cerebrale atrofie
symptoom of symptoomcomplex is er een diffe-
ontstaat. Deze asymmetrie wordt bij circa 50-80%
rentie¨le diagnose op te maken. Bij op de voorgrond
van de CBD-patie¨nten gevonden en is vooral in het
staand parkinsonisme kan er gedacht worden aan
posterieure deel van de frontaal cortex en parie¨taal
idiopathisch of secundair parkinsonisme. De
cortex duidelijk aanwezig in de hersenhelft contra-
grootste differentieeldiagnostische moeilijkheden
lateraal aan de klinisch meest aangedane lichaams-
ontstaan met PSP. Bij op de voorgrond staande
helft. Tegelijkertijd kunnen ook meer subcorticale
oculomotorische stoornissen in combinatie met
afwijkingen in de witte stof worden gezien.
balansproblemen, vallen en een symmetrische bra-
dykinetisch rigidesyndroom, zal de diagnose PSP
j
26.4.2
Nucleair geneeskundig onderzoek
eerder worden gesteld. Bij differentieeldiagnosti-
sche twijfel kan aanvullend onderzoek in de vorm
In het beginstadium van de aandoening bestaat er
van FDG-PET, EEG (Focal Sharp Waves), en moge-
veel differentieeldiagnostische twijfel met andere
lijk ook liquoronderzoek (tau > 180 pg/ml) helpen.
neurodegeneratieve aandoeningen, zoals progres-
Indien frontaalcognitieve verschijnselen in het
sieve supranucleaire blikverlamming (PSP), de
begin meer op de voorgrond blijken te staan, kan
ziekte van Parkinson en multisysteematrofie
gedacht worden aan frontotemporale dementie,
(MSA). Eerder dan er structurele afwijkingen aan
frontaalkwabdementie of primair progressieve
het brein worden gevonden kan middels PET of
afasie. Ook hier kan FDG-PET een mogelijk uit-
SPECT enige twijfel worden weggenomen. Ver-
komst bieden om de differentie¨le diagnoses te
schillende studies hebben een asymmetrisch hy-
verengen (zie tabel 26.3).
pometabolisme van de hemisfeer contralateraal
aan de meest aangedane lichaamshelft aangetoond
(zie figuur 26.1). De aangedane hemisfeer toont
j
26.4
Aanvullend onderzoek
vooral in het striatum en parie¨tale cortex een ver-
minderd metabolisme. Bij PSP is er vooral sprake
Alleen postmortaal onderzoek kan in combinatie
van een bilateraal mediofrontaal en anterior stria-
met de klinische kenmerken een definitieve diag-
taal hypometabolisme. Recent is in een groot co-
nose CBD leveren. Er zijn geen pathognomische
hort van patie¨nten met een bewegingsstoornis de

26 Corticobasale degeneratie
281
meerwaarde van fluorodeoxy-glucose (FDG) PET
interpieklatenties. De in de literatuur veelbespro-
bij de initie¨le diagnose van bewegingsstoornissen
ken ‘giant’ N20 wordt zelden gevonden. Het aan-
aangetoond. Circa 90,9% van de middels FDG-PET
tonen van een verlengde N13-N20 interpieklatentie
aangetoond CBD-patroon bleek bij klinisch ver-
kan wel faciliteren bij het maken van een onder-
volg twee jaar later ook daadwerkelijk een passend
scheid tussen CBD en PSP18.
klinisch profiel te ontwikkelen.
j
26.4.4
Neuropsychologie
j
26.4.3
Klinische neurofysiologie
Systematisch neuropsychologisch onderzoek naar
Het elektro-encefalogram (EEG) bij CBD kan in het
de afwijkingen bij CBD is niet vaak verricht. Pillon
begin normaal zijn, maar zal in de loop van de
en collega’s hebben aangetoond dat er vooral
ziekte gegeneraliseerde of soms asymmetrische
stoornissen in aandacht/concentratie, executieve
vertraging laten zien, vooral aan de zijde contrala-
functies, verbale ‘fluency’, praxis, taal en visuo-
teraal van de klinisch meest aangedane zijde. In
spatie¨le functies bestaan. Dit profiel vertoont grote
vergelijking met andere neurodegeneratieve aan-
gelijkenis met dat bij PSP-patie¨nten. De CBD-
doeningen zijn dit vrij aspecifieke bevindingen.
patie¨nt is echter duidelijk meer aprakisch en heeft
Focal Slow Waves vooral frontaal en parie¨taal ge-
meer moeite met het programmeren van zijn be-
lokaliseerd, kunnen bij diagnostische twijfel tus-
wegingen.
sen CBD en PSP met een positief voorspellende
waarde van 80,0% en negatief voorspellende waar-
j
26.4.5
Psychiatrische verschijnselen
de van 85,7% van meerwaarde zijn.
Somatosensorische evoked potentials hebben te-
Psychiatrische kenmerken als depressie, apathie en
gengestelde resultaten gevonden in absolute en
frontale ontremmingsverschijnselen (bijvoorbeeld
Figuur 26.1
FDG-PET van het cerebrum met opvallend asymmetrisch hypometabolisme van de linker laterofrontale, parie¨tale en temporale cortex, evenals een hypometabolisme van het linker striatum en thalamus bij een patie
¨nt met CBD en
dominerend rechtszijdige klachten.
Handboek dementie
agitatie) komen voor bij 87% van de CBD-patie¨n-
titeiten zijn, die tot het corticobasale syndroom
ten. De combinatie van depressie en agitatie in
kunnen leiden.
afwezigheid van apathie, differentieert met een
CBD behoort tot de zogenaamde tauopathiee¨n,
redelijke accuratesse (88%) CBD van PSP. Vooral
waarbij afwijkingen in het tauproteı¨ne op basis
apathie wordt eerder bij PSP- dan CBD-patie¨nten
van genmutaties een belangrijke rol spelen. Het
gevonden. Visuele hallucinaties of psychoses in
samenspel van tauproteı¨ne en ubiquitine is be-
engere zin zijn niet eerder beschreven en indien
langrijk voor een goed functionerend cytoskelet.
aanwezig dienen deze ter differentie¨ring met bij-
Het afwijkende tauproteı¨ne zal uiteindelijk tot af-
voorbeeld parkinsondementie of diffuse Lewy-
wijkende eiwitdegradering en tot pathologische
body-dementie.
ophoping van niet-verwerkbaar materiaal (zoals
inclusielichaampjes) leiden. Deze prominente tau-
j
26.4.6
Overige
pathologie, associatie met het H1-tau-haplotype, de
aanwezigheid van een ‘four-repeat’ tau-isoform en
De liquor cerebrospinalis toont bij CBD-patie¨nten
overlappende klinische kenmerken, zijn aanwij-
een verhoogd tauproteı¨ne. Het is een aspecifieke
zingen voor gedeelde pathofysiologie bij CBD en
bevinding, omdat dit ook bij andere neurodegene-
PSP. Dit wordt verder ondersteund door het gege-
ratieve aandoeningen en vooral bij andere tauopa-
ven dat bij obductie 50% van de klinisch gediag-
thiee¨n kan worden gevonden. Een verhoogd tau-
nosticeerde CBD-patie¨nten een uiteindelijke diag-
proteı¨ne (cut-off van 180 pg/ml; >180 suggestief
nose PSP krijgen.
voor CBD en <180 suggestief voor PSP) kan wel
In 2002 zijn bovenvermelde neuropathologische
faciliteren bij het differentie¨ren tussen CBD en
kenmerken opnieuw onderzocht en samengevat.
PSP. De toekomst zal moeten uitwijzen of een ver-
Hieruit zijn pathologische criteria voor CBD ge-
hoogd tauproteı¨ne in combinatie met een normaal
postuleerd, namelijk aanwezigheid van corticale
‘neurofilament heavy-chain’ NfHMSI35 een nauw-
en striatale taupositieve neuronale en gliale laesies
keuriger onderscheid kan maken.
(vooral astrocytaire plaques en ‘thread-like’ laesies)
met focaal neuronaal celverlies in de cortex en
substantia nigra.
j
26.5
Neuropathologie bij CBD
De oorzaak van CBD als separate entiteit is niet
j
26.6
Behandeling
bekend. Anderzijds kan het corticobasale syn-
droom veroorzaakt worden door verschillende an-
j
26.6.1
Medicamenteus
dere nosologische entiteiten, zoals frontotempora-
le dementie (FTD), primair progressieve afasie
Een curatieve behandeling ontbreekt en ook effec-
(PPA) en de ziekte van Alzheimer.
tieve symptomatische behandelingen zijn niet of
Macroscopisch staan de frontoparie¨tale corticale
nauwelijks voor CBD voorhanden. Vooral voor de
atrofie, vooral in de perirolandische gebieden, en
corticale disfuncties bestaan er geen effectieve the-
de degeneratie van de substantia nigra bij CBD
rapeutische mogelijkheden.
voorop. Hierbij correleert de corticale atrofie bijna
In de beperkte literatuur over therapeutische
altijd met de lateralisatie van de klinische sympto-
mogelijkheden is het parkinsonisme het meest be-
men. Op hersenstamniveau kan er atrofie van de
schreven. Kompoliti en collega’s hebben in 33 van
tractus corticospinalis worden gevonden. Betrok-
de 128 CBD-patie¨nten (26%) een levodopa respons
kenheid van de temporale cortex wordt vaker ge-
geconstateerd. De klinische relevantie van deze
zien bij andere onderliggende aandoeningen als
respons is zeer beperkt en er is een klein risico van
FTD, PPA en Alzheimer.
door levodopa-geı¨nduceerde dyskinesiee¨n.
Microscopisch is er naast algemeen neuronver-
Andere symptomen, zoals myoclonus en depres-
lies en gliosis nog een ander opvallend fenomeen,
sie kunnen met respectievelijk clonazepam en
namelijk gezwollen ballonvormige achromatische
antidepressiva worden behandeld. Van het gebruik
neuronen in vooral de diepere lagen (in de laminae
van benzodiazepinen bij CBD is een verbetering
III en V) van de gedegenereerde frontoparie¨tale
van parkinsonisme en dystonie beschreven.
cortex. Deze achromatische neuronen zijn de
meest typische cellulaire afwijkingen bij CBD. Bo-
j
26.6.2
Ondersteunend en palliatief
vendien bestaan er net zoveel andere pathologische
afwijkingen als er onderliggende nosologische en-
Belangrijker dan de medicamenteuze opties zijn de
ondersteunende en palliatieve therapiee¨n, zoals
283
het gebruik van een rollator/rolstoel bij loop- en
differential diagnosis of parkinsonian disorders.
balansstoornissen, logopedie voor spraak- en slik-
NeuroImage. 2005;26(3):912-21.
stoornissen, artificie¨le tranen voor conjunctivitis
Houlden H, Baker M, Morris HR, et al. Corticobasal
door verminderde oogknipperfrequentie, anticho-
degeneration and progressive supranuclear palsy
linergica òf focale radiotherapie voor sialorroe, en
share a common tau haplotype. Neurology. 2001;
multidisciplinaire transmurale zorg voor patie¨nt
56(12):1702-6.
en familie.
Josephs KA, Whitwell JL, Dickson DW, et al. Voxel-
based morphometry in autopsy proven PSP and
CBD. Neurobiology of Aging. 2008;29(2):280-9.
j
26.7
Prognose
Kompoliti K, Goetz CG, Boeve BF, et al. Clinical pre-
sentation and pharmacological therapy in cortico-
CBD is een langzaam progressieve aandoening met
basal degeneration. Arch Neurol. 1998;55(7):957-61.
een mediane overleving van 7,9 jaar (met een
Koyama M, Yagishita A, Nakata Y, et al. Imaging of
spreiding van 2,5 tot 12,5 jaar) na de eerste klini-
corticobasal degeneration syndrome. Neuroradiolo-
sche presentatie van de aandoening. Het eerste be-
gy. 2007;49(11):905-12.
zoek aan de neuroloog is meestal 3 jaar na de eerste
Litvan I, Cummings JL, Mega M. Neuropsychiatric
klinische symptomen, waardoor bij het stellen van
features of corticobasal degeneration. J Neurol
de diagnose de mediane overleving circa 5 jaar be-
Neurosurg Psychiatry. 1998;65(5):717-21.
draagt. Een aspiratiepneumonie is de meest fre-
Monza D, Ciano C, Scaioli V, et al. Neurophysiological
quente oorzaak van overlijden.
features in relation to clinical signs in clinically
diagnosed corticobasal degeneration. Neurol Sci.
2003;24(1):16-23.
Literatuur
Nagahama Y, Fukuyama H, Turjanski N, et al. Cere-
bral glucose metabolism in corticobasal degenera-
Ay H, Buonanno FS, Price BH, et al. Sensory alien
tion: comparison with progressive supranuclear
hand syndrome: case report and review of the lite-
palsy and normal controls. Mov Disord. 1997;12(5):
rature. J Neurol Neurosurg Psychiatry. 1998;65(3):
691-6.
366-9.
Okuda B, Tachibana H, Takeda M, et al. Asymmetric
Blin J, Vidailhet MJ, Pillon B, et al. Corticobasal
changes in somatosensory evoked potentials corre-
degeneration: decreased and asymmetrical glucose
late with limb apraxia in corticobasal degeneration.
consumption as studied with PET. Mov Disord.
Acta Neurol Scand. 1998;97(6):409-12.
1992;7(4):348-54.
Pezzoli G, Canesi M, Galli C. An overview of parkin-
Boeve BF, Lang AE, Litvan I. Corticobasal degenera-
sonian syndromes: data from the literature and
tion and its relationship to progressive supranu-
from an Italian data-base. Sleep Med. 2004;5(2):
clear palsy and frontotemporal dementia. Ann
181-7.
Neurol. 2003;54 Suppl 5:S15-9.
Pillon B, Blin J, Vidailhet M, et al. The neuropsycho-
Boxer AL, Geschwind MD, Belfor N, et al. Patterns of
logical pattern of corticobasal degeneration: com-
Brain Atrophy That Differentiate Corticobasal
parison with progressive supranuclear palsy and
Degeneration Syndrome From Progressive Supra-
Alzheimer’s disease. Neurology. 1995;45(8):1477-83.
nuclear Palsy. Arch Neurol. 2006;63(1):81-6.
Rebeiz JJ, Kolodny EH, Richardson EP, Jr. Cortico-
Brettschneider J, Petzold A, Sussmuth SD, et al. Neu-
dentatonigral degeneration with neuronal achro-
rofilament heavy-chain NfH(SMI35) in cerebrospi-
masia. Arch Neurol. 1968;18(1):20-33.
nal fluid supports the differential diagnosis of Par-
Schneider JA, Watts RL, Gearing M, et al. Corticobasal
kinsonian syndromes. Mov Disord. 2006;21(12):
degeneration: neuropathologic and clinical hetero-
2224-7.
geneity. Neurology. 1997;48(4):959-69.
Dickson DW, Bergeron C, Chin SS, et al. Office of Rare
Takeda M, Tachibana H, Okuda B, et al. Electrophy-
Diseases neuropathologic criteria for corticobasal
siological comparison between corticobasal degene-
degeneration. J Neuropathol Exp Neurol. 2002;
ration and progressive supranuclear palsy. Clin
61(11):935-46.
Neurol Neurosurg. 1998;100(2):94-8.
Doi T, Iwasa K, Makifuchi T, et al. White matter
Taniwaki T, Yamada T, Yoshida T, et al. Heteroge-
hyperintensities on MRI in a patient with cortico-
neity of glucose metabolism in corticobasal dege-
basal degeneration. Acta Neurol Scand. 1999;99(3):
neration. J Neurol Sci. 1998;161(1):70-6.
199-201.
Tashiro K, Ogata K, Goto Y, et al. EEG findings in
Eckert T, Barnes A, Dhawan V, et al. FDG PET in the
early-stage corticobasal degeneration and progres-
sive supranuclear palsy: a retrospective study and
Handboek dementie
literature review. Clin Neurophysiol. 2006;117(10):
patients with corticobasal degeneration or progres-
2236-42.
sive supranuclear palsy. J Neurol Sci. 2001;183(1):
Togaski DM, Tanner CM. Epidemiologic aspects. In:
95-8.
Litvan I, Goetz C, Lang AE, editors. Corticobasal
Wenning GK, Litvan I, Jankovic J, et al. Natural his-
degeneration and related disorders. London: Lip-
tory and survival of 14 patients with corticobasal
pincott Williams and Wilkins;. 2000. pp. 53-60.
degeneration confirmed at postmortem examina-
Urakami K, Wada K, Arai H, et al. Diagnostic signifi-
tion. J Neurol Neurosurg Psychiatry. 1998;64(2):
cance of tau protein in cerebrospinal fluid from
84-9.
14-3-3-test
265
Alzheimer Disease Assessment Scale-cogni-
15-woordentest
35
tieve sectie (ADAS-cog)
61
Alzheimer, ziekte van
38, 45
A
206
–, agnosie
200
aandacht
–, alcoholgebruik
249
–, selectief
36
–, apraxie
200
–, volgehouden
36
–, bloedonderzoek
202
aandachtsfunctie
30
–, CT
203
aandachtsstoornissen
30
–, farmacologie
208
ABC-model
141
–, geheugenstoornis
200
a-bèta
205
–, genetisch aspect
207
abstractievermogen
32
–, gerstmannsyndroom
200
additionele diagnostiek
116
–, klassiek
198
ADL, beoordelingsschaal
65
–, liquor cerebrospinalis
202
advance care planning
164, 170
–, macroscopie
203
afasie, primair progressief
39
–, MCI
192
afzondering
183
–, microscopie
203
agitatie
145
–, MRI
202
–, overige middelen
147
–, neuropsychiatrie
201
agressie
145
–, neuropsychologie
201
–, antipsychoticum
145
–, pathogenese
206
–, overige middelen
147
–, PET
203
alcoholgebruik
135
–, preseniel
198
–, behandeling
249
–, preventie
208
–, cognitieve stoornis
244
–, prosopagnosie
200
–, dementie
245
–, seniele variant
198
–, frontotemporale dementie
248
–, somatiek
201
–, genetisch aspect
249
–, taalstoornis
200
–, MRI
247
–, tongapraxie
200
–, neuropathologie
249
–, uitvoerende functiestoornis
201
–, neuropsychologisch onderzoek
247
–, visuele agnosie
200
–, preventie
250
Alzheimercafe´
157
–, vasculaire dementie
249
amnestische MCI
189
–, ziekte van Alzheimer
249
amyloı¨d precursor proteı¨ne (APP)
206, 227
alcoholgerelateerde dementie
245
anamnese
26, 34
alcoholic dementia
245
angst
148
alcoholonttrekkingsdelier
247
anti-amyloı¨d behandeling
129
alfasynucleı¨negen, mutatie
239
anticipatie
256
alfatocoferol
130
antilichaam Creutzfeldt-Jakob
266
alien limb
278
antioxidantia
130
Alzheimer
8, 197
antiparkinsongeneesmiddelen
258
antipsychoticum
Handboek dementie
–, agressie
145
–, mentorschap
177
–, verontrustende gegevens
146
beslissingsbekwaamheid
164
–, werkzaamheid
146
beslisvaardigheid
164
apathie
148, 223
bèta-amyloı¨d
205
Apathie Evaluation Scale (AES)
64
bèta-amyloı¨d precursor-gen (APP)
207
Apathy Scale (AS)
64
bèta-vouwbladstructuur
266
APOE-gen
208
bewegingsstoornis, corticobasale degenera-
apraxie
31
tie
278
–, corticobasale degeneratie
278
Binswanger-dementie
221
aromatherapie
140
biomarker
10
ataxie
262
biopt Creutzfeldt-Jakob
266
atrofie, posterieure corticale
39
bloedonderzoek, parkinsondementie
235
autonomie
162
BOLD-effect
45
–, respect voor
163
bovine spongiform encephalopathy (BSE)
263
BSE, zie bovine spongiform encephalopathy
Balint, syndroom van
29, 31
bed-side-test
30
CAA, zie congofiele cerebrale angiopathie
beeldvormend onderzoek
CADASIL, zie subcortical infarctions and
–, corticobasale degeneratie
280
leucoencephalopathie
–, frontotemporale dementie
215
CAG-repeat
256
–, parkinsondementie
234
Cambridge Examination for Mental Disor-
–, vasculaire dementie
225
ders, Revised (CAMCOG-R)
61
behandeling
casemanagementprogramma
107
–, alcoholgebruik
249
casemanager
110
–, alfatocoferol
130
CBD, zie corticobasale degeneratie
–, anti-amyloı¨d
129
CBO-richtlijn
107
–, antioxidantia
130
cerebellair verschijnsel
263
–, cholinesterase-inhibitor
130
cerebrale veroudering
8
–, cholinesteraseremmer
130
ChE-I, zie cholinesterase-inhibitor
–, donepezil
130
cholinesterase-inhibitor (ChE-I)
130
–, ethische aspecten
132
cholinesteraseremmer
116, 130
–, galantamine
130
circumlocutie
31
–, immunisatietherapie
129
Clinical Dementia Rating Scale (CDR)
62
–, indicatie
133
CLSM-zorg
120
–, Lewy-body-dementie
240
CMAI, zie Cohen-Mansfield Agitation Inven-
–, memantine
131
tory
–, patie¨nt en familie
132
codon-129-polymorfisme
267
–, preventief
134
cognitieve functie
–, rivastigmine
130
–, aandacht
36
–, selegeline
130
–, betrouwbaarheid tests
36
–, tacrine
130
–, concentratie
36
belevingsgerichte orie¨ntatie
163
–, executieve functies
36
belevingsgerichte zorg
139
–, geheugen
35
belevingsperspectief
163
–, taal
35
beoordelingsschaal
–, visueel-constructief
36
–, ADL
65
–, visueel-ruimtelijk
36
–, delier
63
cognitieve functiestoornis, typering
37
–, IADL
65
Cognitieve screeningstest (CST)
60
–, Likert-type
62
cognitieve stimulatie
140
–, visueel analoge schaal (VAS)
62
cognitieve stoornis
–, zelfbeoordeling
62
–, alcoholgebruik
244
beschermingsbewind
177
–, zonder dementie
28
beschermingsmaatregel
176
cognitieve therapie, MCI
195
–, beschermingsbewind
177
cognitieve training
135
–, curatele
176
287
Cohen-Mansfield Agitation Inventory
de-institutionalisering
125
(CMAI)
64
delier
28, 123
communicatieve ethiek
163
–, beoordelingsschaal
63
compulsief gedrag
148
Dementia Quality of Life instrument (DQoL)
65
computertomografie (CT)
43, 266
dementie
7
confabuleren
246
–, alcoholgerelateerde
245
congofiele cerebrale angiopathie (CAA)
206
–, definitie
7
consensuscriterium, progressieve supranu-
–, demografische factor
16
cleaire verlamming
270
–, diagnose
10
Cornell Scale for Depression in Dementia
–, ernstige
27
(CSDD)
64
–, frontotemporale
38, 55
corticaal sensibele stoornissen
278
–, genetische factor
16
corticale functiestoornis
263
–, incidentie
14
corticobasale degeneratie
40, 50
–, kosten
20
–, apraxie
278
–, lichte
27
–, beeldvormend onderzoek
280
–, lifetimerisico
15
–, bewegingsstoornis
278
–, mantelzorg
138
–, farmacotherapie
282
–, matig ernstige
27
–, functiestoornis
278
–, mengbeeld
14
–, klinische neurofysiologie
281
–, multi-infarct
40
–, neuropathologie
282
–, neurodegeneratieve aandoening
37
–, neuropsychologie
281
–, omgevingsfactor
17
–, nucleair geneeskundig onderzoek
280
–, Parkinson
39
–, palliatieve therapie
282
–, post-stroke
40
–, postmortaal onderzoek
280
–, risicofactor
15
–, prognose
283
–, semantische
39
–, psychiatrisch verschijnsel
281
–, seniele
8
corticobasale degeneratie (CBD)
40, 50
–, strategisch infarct
40
Creutzfeldt-Jakob
–, subcorticaal ischemisch vasculair
40
–, 14-3-3-test
265
–, subtypen
13
–, antilichaam
266
–, terminale aandoening
162
–, biopt
266
–, vasculair
40
–, CT
266
–, vasculaire
47, 55
–, EEG
56, 265
–, verlies van zelfbepaling
161
–, farmacotherapie
267
–, verpleeghuis
121
–, fysieke problemen
263
–, verwachte groei
20
–, genetisch aspect
267
–, wilsbekwaam
161
–, iatrogeen
264
–, wilsonbekwaam
162
–, MR
266
dementie-decompensatie
124
–, preventie
267
dementiesyndroom
29, 38
–, psychiatrisch symptoom
262
–, differentie¨le diagnose
27
–, SPECT
266
–, incidentie
11
–, symptoomfrequentie
263
–, klinisch onderzoek
29
–, therapie
263
–, lichamelijk onderzoek
32
–, visusafwijking
263
dementiezorg
11
–, ziekteverschijnsel
262
demografische factor dementie
16
CSDD, zie Cornell Scale for Depression in
depressie
148, 223
Dementia
–, geheugenklachten
28
CST, zie cognitieve screeningstest
–, primair
28
CT, zie computertomography
Depressielijst
64
curatele
176
depressieve dementie
28
diabetes mellitus
134
declaratief geheugen
245
diagnose dementie
10
decubitus
122
diagnose per exclusionem
201
definitie MCI
189
Handboek dementie
Diagnostic and statistical manual of mental
ernstige dementie
27
disorders 4th edition text revised (DSM-IV-
ERP, zie event-related potential
TR)
10
Ervaren Druk door Informele Zorg (EDIZ)
65
diagnostiek
ethiek
–, additioneel
116
–, communicatief
163
–, etiologisch
116
–, interactioneel
163
–, inclusief
201
etiologische diagnose
116
–, nosologisch
25
euthanasie
–, syndromaal
25, 116
–, gezamenlijkheid
170
differentie¨le diagnose dementiesyndroom
27
–, moreel bezwaar
169
differentie¨le diagnose vasculaire dementie
224
–, samenspraak
170
disease management program (DMP)
107
–, zorgvuldigheidsvoorwaarde
169
diseasemanagement
107
euthanasieverklaring
169
DLB, zie Lewy-body-dementie
event-related potential (ERP)
57
DMP, zie disease management program
evoked potential (EP)
57
DNA-diagnostiek
216
excess disability
163
donepezil
130
executieve functie
31
Down, syndroom van
20
expertteam
111
DQoL, zie Dementia Quality of Life instru-
extramurale verpleeghuiszorg
125
ment
extrapiramidaal symptoom
224
DSM-IV-TR
10
extrapiramidaal verschijnsel
263
dwangbehandeling
183
dwanghuilen
148
FAB, zie Frontal Assessment Battery
dyade patie¨nt-mantelzorger
141
Fabry, ziekte van
227
dysartrie
271
farmacotherapie
–, corticobasale degeneratie
282
EDIZ, zie Ervaren Druk door Informele Zorg
–, Creutzfeldt-Jakob
267
EEG
53
–, frontotemporale dementie
217
–, antidepressiva
57
–, MCI
193
–, antipsychotica
57
–, vasculaire dementie
227
–, beperking
53
–, ziekte van Alzheimer
208
–, corticobasale degeneratie
56
–, ziekte van Huntington
258
–, Creutzfeldt-Jakob
265
FAST, zie Functional Assessment Staging
–, delier
57
Fatale Familiaire Insomnie (FFI)
264
–, epilepsie
57
fenokopie
257
–, frontotemporale dementie
55
fenotype
272
–, indicatie
54
FFI, zie Fatale Familiaire Insomnie
–, Lewy-body-dementie (DLB)
56
fixatie
123
–, parkinsondementie
55, 234
fMRI
45
–, progressieve supranucleaire parese
56
focale neuropsychologische stoornis
29
–, toxische encefalopathie
57
fonologische fluency
35
–, vasculaire dementie
55
fragmentatie gezondheidszorg
108
–, voordeel
53
Frontal Assessment Battery (FAB)
61, 273
–, ziekte van Alzheimer
54
frontotemporale dementie (FTD)
38
–, ziekte van Creutzfeldt-Jakob
56
–, alcoholgebruik
248
–, ziekte van Huntington
56
–, apathie
213
eetstoornis
148
–, beeldvormend onderzoek
215
elektro-encefalogram (EEG)
53, 190
–, EEG
55
emotionele labiliteit
148, 223
–, emotionele onverschilligheid
213
EP, zie evoked potential (EP)
–, farmacotherapie
217
episodisch geheugen
31
–, genetische aspecten
216
episodische geheugentest
222
–, heteroanamnese
212
erfelijke vorm prionziekte
264, 266
–, immunohistochemie
216
ERGO-onderzoek
15
–, impulsiviteit
213
ergotherapie
141
–, initiatiefverlies
213
289
–, liquoronderzoek
216
–, monogenetisch
16
–, neurofysiologisch onderzoek
215
–, susceptibiliteitgen
17
–, neuropathologisch onderzoek
216
gen-gen-interactie
20
–, neuropsychologisch onderzoek
214
genome wide association (GWA)
17
–, obsessief-compulsief gedrag
213
gen-omgevingsinteractie
20
–, ontremd gedrag
213
Geriatric Depression Scale (GDS)
64
–, preventie
217
Gerstmann, syndroom van
31
–, spraakprobleem
213
Gerstmann-Straüssler-Scheinkersyndroom
–, taalprobleem
213
(GSS)
264
–, ziekte-inzicht
212
gezondheidszorg, fragmentatie
108
frontotemporale lobaire degeneratie
47
Ginkgo-extract
194
FTD, zie frontotemporale dementie
GIP, zie Gedragsobservatieschaal voor de
functiestoornis, corticobasale degeneratie
278
Intramurale Psychogeriatrie
Functional Assessment Staging (FAST)
63
Global Deterioration Scale (GDS)
63, 121
functionele kernspintomografie (fMRI)
45
GSS, zie Gerstmann-Straüssler-Scheinkersyn-
fysieke problemen Creutzfeldt-Jakob
263
droom
GWA, zie genome wide association
galantamine
130
GDS, zie Geriatric Depression Scale
hallucinatie
147
Gedragsobservatieschaal voor de Intramurale
head turning sign
30
Psychogeriatrie (GIP)
63
hersenen, pathologisch proces
9
gedragsprobleem
hersenpathologie
7
–, beloop
144
hersenreserve
9
–, farmacologie
144
heteroanamnese
26
–, medicatie off label
144
–, frontotemporale dementie
212
–, probleemanalyse
145
–, MCI
189
–, psychosociale factor
144
hubmodel
106
–, terminologie
143
huisarts
105
–, voorkomen
144
–, mantelzorger
106
gedragstherapie
141
–, multidisciplinaire aanpak
106
gedragsverandering, ziekte van Huntington
255
–, poortwachter
117
geheugenklachten bij depressie
28
Huntington, ziekte van
geheugenpolikliniek
–, cognitieve stoornis
255
–, additionele diagnostiek
116
–, EEG
56
–, diagnose
116
–, executieve stoornis
255
–, etiologische diagnose
116
–, farmacotherapie
258
–, farmacologie
116
–, gedragsverandering
255
–, non-farmacologie
116
–, genetisch aspect
256
–, onderzoek
116
–, juveniele vorm
256
–, ontwikkeling
114
–, motorisch verschijnsel
254
–, procedure
114
–, neuropathologie
257
–, verwijzing
117
–, obsessief-compulsieve verschijnselen
255
genetisch aspect
–, PCR
256
–, alcoholgebruik
249
–, preventie
258
–, Creutzfeldt-Jakob
267
–, psychiatrisch probleem
255
–, frontotemporale dementie
216
–, stemmingsstoornis
255
–, Lewy-body-dementie
239
–, suı¨cide
255
–, MCI
191
–, ziektebeloop
256
–, parkinsondementie
239, 240
hyperfagie
122
–, progressieve supranucleaire verlam-
hyperlipidemie
135
ming
274
hypertensie
134
–, vasculaire dementie
227
hypoperfusie
221
–, ziekte van Alzheimer
207
–, ziekte van Huntington
256
IADL, beoordelingsschaal
65
genetische factor dementie
iatrogene Creutzfeldt-Jakob
264
Handboek dementie
immunisatietherapie
129
Lewy-insluitlichaampjes
235
–, Bapineuzumab
130
Lewy-pathologie
239
immunohistochemie, frontotemporale
lichaamsbeweging
135
dementie
216
lichamelijk onderzoek
32
incidentie dementiesyndroom
11
lichte dementie
27
inclusieve diagnostiek
201
lichttherapie
140
Informant Questionnaire on Cognitive
Likert-type, beoordelingsschaal
62
Decline in the Elderly (IQCODE)
63
liquoronderzoek, parkinsondementie
235
informed consent
162
LRRK2-gen, mutatie
239
intakefase
114
lumbaalpunctie
191
integratie van zorg
108
interactionele ethiek
163
magnetic resonance imaging (MRI)
44
intercurrente morbiditeit
122
–, alcoholgebruik
247
intermitterend breinfalen bij dementie
124
–, progressieve supranucleaire verlam-
interventie
ming
272
–, aromatherapie
140
magnetische resonantie (MR)
266
–, cognitieve stimulatie
140
magneto-encefalografie (MEC)
57
–, ergotherapie
141
mantelzorger
140
–, gedragstherapie
141
–, ABC-model
141
–, lichttherapie
140
–, competentie
141
–, mantelzorger
140
–, counseling
158
–, muziektherapie
139
–, draagkracht
107
–, psychomotorische therapie
140
–, draaglast
107
–, reminiscentietherapie
139
–, elektronische ondersteuning
158
–, snoezelen
139
–, huisarts
106
interventiekeuze
–, ondersteuningsproject
107
–, hulpverlener
138
–, psycho-educatie
157
–, sociale context
138
–, psychotherapie
158
IQCODE, zie Informant Questionnaire on
–, voorlichting
156
Cognitive Decline in the Elderly
Marchiafava Bignami
247
ischemische grotevaatletsels
221
matig ernstige dementie
27
MCI, zie mild cognitive impairment
ketenmodel
106
MEC, zie magneto-encefalografie
klaptest
273
medicatie
klinisch onderzoek dementiesyndroom
29
–, ethische aspecten
132
korsakovsyndroom
245
–, off label
144
kuru
265
memantine
131
kwaliteit van leven
171
mentorschap
177, 178
metabolisme PET-scan
191
laboratoriumonderzoek, MCI
190
middelen en maatregelen
183
letselpreventie
123
mild cognitive impairment
levensverlenging
172
–, amnestisch
189
leverfalen
244
–, cognitieve therapie
195
levitation
278
–, definitie
189
Lewy-body-dementie (DLB)
40, 48
–, diagnose
192
–, behandeling
240
–, farmacotherapie
193
–, cognitie
241
–, genetisch aspect
191
–, consensuscriterium
235
–, heteroanamnese
189
–, EEG
56
–, laboratoriumonderzoek
190
–, fluctuatie klinisch beeld
237
–, neuropsychologisch onderzoek
190
–, genetisch aspect
239
–, non-amnestisch
189
–, motorisch verschijnsel
235
–, oorzaak
191
–, neuropathologische verandering
238
–, prevalentie
189
–, neuropsychologisch onderzoek
238
–, preventie
193
–, visuele hallucinatie
237
–, psychiatrisch symptoom
189
291
–, reversibiliteit
189
non-interferentie
162
–, ziekte van Alzheimer
192
noo¨tropicum
194
–, ziekteverloop
189
normaal prioneiwit
266
mild cognitive impairment (MCI)
38, 132
normaal priongen
267
Mini Mental State Examination (MMSE)
60
normale veroudering
38
Montreal Cognitive Assessment (MoCA)
61
normatief ethisch vraagstuk
164
morele verantwoordelijkheid
169
nosologische benadering
10
motorisch verschijnsel
nosologische diagnostiek
10, 25
–, progressieve supranucleaire verlam-
NPO, zie neuropsychologisch onderzoek
33
ming
270
–, ziekte van Huntington
254
observatie
30
motorisch voorhoornlijden
214
–, non-verbaal gedrag
34
MRI, zie magnetic resonance imaging
–, verbaal gedrag
34
MSA, zie multisysteematrofie
obstructief slaapapnoesyndroom (OSAS)
54
multidisciplinaire aanpak, huisarts
106
off label medicatie
144
multi-infarct dementie
40
omgevingsfactor dementie
multimorbiditeit
11, 108
–, alcohol
19
multisysteematrofie (MSA)
40, 50
–, antioxidant
19
muziektherapie
139
–, cerebrale vaatschade
18
myoclonus
262
–, cholesterol
18
–, diabetes
18
neglect
31
–, fysieke activiteit
19
neuritische plaque
205
–, homocysteı¨ne
18
neuroimaging
190
–, hormonen
18
neuronale degeneratie
257
–, hypertensie
17
neuropathologie
–, medicatie
19
–, alcoholgebruik
249
–, mentale activiteit
19
–, corticobasale degeneratie
282
–, NSAID
19
–, frontotemporale dementie
216
–, obesitas
18
–, Lewy-body-dementie
238
–, roken
18
–, parkinsondementie
238
–, sociale activiteit
19
–, progressieve supranucleaire verlam-
–, statine
19
ming
273
omgevingsinterventie
124
–, vasculaire dementie
226
ondervoeding
122
–, ziekte van Huntington
257
onderzoek cognitief domein
30
Neuropsychiatric Inventory (NIP)
63
onderzoek mentale status
29
neuropsychiatrische symptomen
124
on-off-verschijnsel
240
neuropsychologie
ontremming
149
–, corticobasale degeneratie
281
opnameduur verpleeghuis
120
–, progressieve supranucleaire verlam-
ouderdomsvergeetachtigheid
27
ming
273
overgewicht
134
neuropsychologisch onderzoek (NPO)
33
–, alcoholgebruik
247
PACSLAC-D
123
–, frontotemporale dementie
214
palliatief zorgconcept
170
–, indicatie
40
parafasie, semantisch
31
–, Lewy-body-dementie
238
Parkinson
232
–, MCI
190
parkinsondementie
50
–, parkinsondementie
234
–, beeldvormend onderzoek
234
niet-inmenging
162
–, bloedonderzoek
235
NINDS-AIREN-criteria
221, 224
–, cognitie
241
NINDS-AIREN-diagnose
222
–, EEG
55, 234
NIP, zie Neuropsychiatric Imaging
–, genetisch aspect
239, 240
non cancer palliative care
170
–, liquoronderzoek
235
non-amnestische MCI
189
–, motorfunctie
240
non-farmacologische interventie
124
–, neuropathologische verandering
238
Handboek dementie
–, neuropsychologisch onderzoek
234
primair progressieve afasie (PPA)
39
–, psychiatrisch verschijnsel
233
primaire depressie
28
–, psychische symptomen
240
primitieve reflex
32
–, risicofactor
233
prioneiwit
266
parkinsondementiecomplex
233
–, BSE
266
parkinsonisme
39
–, kuru
266
Parkinson-plus syndroom
40
–, normaal
266
pathologisch prioneiwit
266
–, pathologisch
266
pathologisch proces in hersenen
9
priongen, normaal
267
pathoplastisch effect
30
prionziekte
PCA, zie posterieure corticale atrofie
–, bèta-vouwbladstructuur
266
pellagra
247
–, erfelijke vorm
264, 266
perfusie SPECT-scan
191
privacy, schending
164
periodic lateralized epileptiform discharges
procerus-teken
271
(PLED’s)
56
progressieve supranucleaire verlamming
periodic short-interval diffuse discharges
–, consensuscriterium
270
(PSIDD’s)
56
–, fenotype
272
perseveratie
32
–, genetisch aspect
274
persoonlijk gemachtigde
181
–, klaptest
273
persoonlijkheidsverandering
223
–, motorisch verschijnsel
270
PET, zie positron emission tomography
–, MRI
272
PET-scan
191
–, neuropathologie
273
piramidaal symptoom
224
–, neuropsychologie
273
piramidale afwijking
263
–, prognose
274
plaque, neuritisch
205
–, spierstoornis
271
PLED’s, zie periodic lateralized epileptiform
progressive non-fluent aphasia (PNFA)
214
discharges
pseudodementie
28
PNFA, zie progressive non-fluent aphasia
PSIDD’s, zie periodic short-interval diffuse
poortwachter, huisarts
117
discharges
positron emission tomography (PET)
45
psychiatrisch symptoom
posterieure corticale atrofie (PCA)
39
–, Creutzfeldt-Jakob
262
post-stroke dementie
40
–, MCI
189
PPA, zie primair progressieve afasie
psychometrisch testonderzoek
34
praxis
31
–, betrouwbaarheid
34
predementie
26
–, stoorfactoren
34
prenataal testen
257
–, testuitslag
34
presymptomatische test
257
–, validiteit
34
prevalentie, MCI
189
psychometrische eisen
34
preventie
134
psychomotorische therapie
140
–, alcoholgebruik
135, 250
–, cognitieve training
135
reminiscentietherapie
139
–, Creutzfeldt-Jakob
267
respijtzorg
158
–, diabetes mellitus
134
response shift
168
–, frontotemporale dementie
217
retrograde amnesie
245
–, hyperlipidemie
135
Revised Memory and Behavioural Problems
–, hypertensie
134
Checklist (RMBPC)
64
–, lichaamsbeweging
135
rivastigmine
130
–, MCI
193
roken
135
–, overgewicht
134
–, roken
135
Schedule for the Evaluation of Individual
–, vasculaire dementie
228
Quality of Life (SEIQoL)
65
–, voeding
135
schriftelijke wilsverklaring
167, 182
–, ziekte van Alzheimer
208
selectieve aandacht
36
–, ziekte van Huntington
258
selegeline
130
prikkelbaarheid
148
semantisch geheugen
31
293
semantische dementie
39, 214
–, neuropathologie
226
semantische fluency
35
–, preventie
228
semantische parafasie
31
–, psychiatrisch symptoom
223
semantische-associatietest
36
–, radiologische aantasting
224
seniele dementie
8
–, risicoprofiel
225
Seven Minute Screen (7MS)
60
verhoogde afleidbaarheid
30
Severe Impairment Battery (SIB)
61
vermaatschappelijking van zorg
125
shared decision-making
163, 167
vermaatschappelijking van zorg, verpleeg-
simultaanagnosie
31
huiszorg
125
single photon emission computerized tomo-
verouderingshypothese
8
graphy (SPECT)
45, 266
verpleeghuis
slaapstoornis
148
–, delier
122
slikproblemen
122
–, materie¨le omgeving
125
snoezelen
139
–, opnameduur
120
SPECT, zie single photon emission computer-
–, restinstituut
126
ized tomography
–, zorgproblemen
121
strategisch infarct dementie
40
verpleeghuisgeneeskunde
120
subcortical infarctions and leuco-
vertegenwoordiger, benoeming
180
encephalopathie (CADASIL)
47, 227
verticale blikverlamming
271
subcorticale dementie
233
vignetmethode
166
subcorticale ischemische vasculaire
visueel analoge schaal (VAS)
62
dementie
40
visuele agnosie
31
sundowning fenomeen
123
visuele extinctie
31
symptomatische therapie
258
visuospatie¨le functie
31
symptoomfrequentie Creutzfeldt-Jakob
263
visusafwijking Creutzfeldt-Jakob
263
syndromale benadering
9
voeding
135
syndromale diagnose
116
volgehouden aandacht
36
syndromale diagnostiek
10, 25
vraaggestuurde zorg
125
syndroom van Balint
29, 31
syndroom van Down
20
waan
147
syndroom van Gerstmann
31
webmodel
106
weigeringsverklaring
168
tacrine
130
wernicke-encefalopathie
245
tangles
206
Wet bopz
182
tauproteı¨ne
206
wettelijk vertegenwoordiger
180
trinucleotide repeat
256
WGBO
181
–, schriftelijke wilsverklaring
182
uitvoerende functie
31
–, uitzondering
182
–, vier soorten vertegenwoordigers
182
valincidenten
123
wijkmeldpunt
110
vallen
wilsbekwaam
164
–, extrinsieke risicofactoren
123
wilsbekwaamheid
–, intrinsieke risicofactoren
123
–, beoordelen
166
valpreventie
123
–, criterium
165
VAS, zie visueel analoge schaal
–, risicoafhankelijkheid
165
vasculaire dementie
40, 47
wilsonbekwaam
175
–, alcoholgebruik
249
–, vaststelling
176
–, beeldvormend onderzoek
225
–, vertegenwoordiger
167
–, differentie¨le diagnose
224
–, volledig
167
–, EEG
55
wilsverklaring
–, farmacotherapie
227
–, dilemma
168
–, geheugenstoornis
222
–, negatief
168
–, genetische aspecten
227
–, positief
168
–, klinisch verloop
223
–, schriftelijk
167
–, neurologisch symptoom
224
Handboek dementie
zaakwaarneming
181
zelfredzaamheid
223
zelfbeoordelingsschaal
62
ziekte-inzicht
163
zelfbinding
169
ziekteverschijnsel Creutzfeldt-Jakob
262
zelfpaternalisme
169
zorgbehoefte
29